
Minor Chapters, Major Impacts - What United States Pharmacopeia Chapters <51>, <61>, <62>, and <1207> mean for your compounding practice
Brian Kelley, BS, ARL Bio Pharma Director of Business Development
International Journal of Pharmaceutical Compounding, Vol. 25, No. 2, March / April 2021, p. 115-124
Abstract
The United States Pharmacopeial Convention, Inc. recommends within the standards of the United States Pharmacopeia that compounding pharmacies have staff dedicated to quality assurance and quality control to ensure patients are receiving safe medications. The quality-control program must include testing. While compounding pharmacies have grown familiar with potency, sterility, and endotoxin testing, there are many more tests recommended within the United States Pharmacopeia that are critical for evaluating the quality of compounded preparations. This article discusses when a few of these tests should be utilized, how to assign acceptance criteria, and how test results are obtained.
Quality Assurance and Quality Control
United States Pharmacopeia (USP) chapters <795> and <797> state that compounding pharmacies should have staff dedicated to quality assurance (QA) and quality control (QC). QA is responsible for procedures, activities, and oversight, while QC is responsible for sampling, testing, and documenting the test results to ensure that specifications have been met before dispensing compounded preparations. USP Chapter <1163> Quality Assurance in Pharmaceutical Compounding is referred to in chapters <795> and <797> because it provides guidance on how to set up QA/QC programs in a compounding pharmacy and makes recommendations for appropriate tests to conduct on compounded preparations. TESTING The goal of testing compounded preparations is to determine the adequacy of the compounding process and the quality of the preparation. Not only is testing the finished preparation recommended in USP Chapter <1163>, but so is testing of intermediates or stock solutions. Testing of stocks is important because the final preparation is likely to be out of specification (OOS) if the stock is not correct. Chapter <1163> also says that compounders must have a basic understanding of pharmaceutical analysis and that test acceptance criteria must be determined prior to testing. The Chapter acknowledges that testing of all preparations is not practical or required, but compounders should know the importance of:
- testing,
- when to test,
- what to test,
- the appropriate test method and equipment,
- how to interpret test results,
- limits of the test, and
- actions to take when a test result is OOS.
Chapter <1163> lists many tests and provides details on the appropriateness of each test. This article focuses on three tests in USP Chapter <1163> and one from the U.S. Food and Drug Administration (FDA) that are not well-known but are important for checking the quality of compounded nonsterile and sterile preparations for release and beyond-use dating (BUD). The tests listed in Chapter <1163> and discussed in this article relate to chapters:
- <61> Microbial Enumeration Tests (applies to non-sterile products)
- <62> Tests for Specified Organisms (a recommended test on nonsterile products)
- <51> Antimicrobial Effectiveness Testing (applies to preserved multi-dose nonsterile and sterile aqueous products)
- <1207> Package Integrity Testing – Sterile Products (applies to sterile and nonsterile preparations)
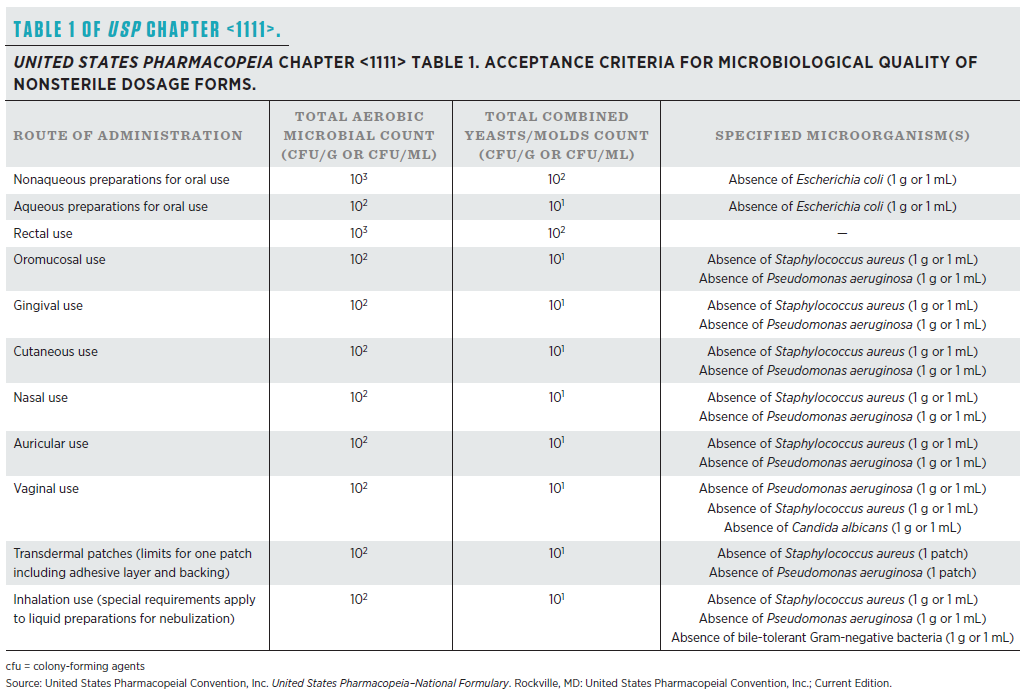
United States Pharmacopeia Chapter <61> Microbial Enumeration Tests
USP <61> applies to nonsterile products, which includes finished nonsterile compounded preparations, and may be used to assess intermediates of sterile compounds. This is a test that determines how many microorganisms are present in nonsterile drug products and sterile products that are made using nonsterile components prior to sterilization of the final preparation. The microbial enumeration test is often referred to as “bioburden” or “microbial limits.” In 483s issued to 503A pharmacies, the FDA has also referred to these tests as “yeast and mold counts” and “presence of microorganisms.” As stated in Chapter <1163>, the acceptance criteria must be defined prior to test initiation. Acceptance criteria are not provided in Chapter <61>. Instead, acceptance criteria recommendations are provided in USP Chapter <1111> or the USP monograph. The limits listed in Table 1 of USP Chapter <1111> list how many microorganisms can be present in nonsterile compounded preparations. Separate criteria are provided for each of the two tests included in USP Chapter <61>.

Table 2 of USP Chapter <1111> provides recommended limits for nonsterile components used to produce sterile preparations. USP Chapter <61> states the compounder must also consider the acceptance criteria of a nonsterile product or component when considering its use, to include:
- the nature of a nonsterile product,
- the method of application,
- the patient,
- the use of immunosuppressive agents/corticosteroids, and
- the presence of disease, wounds, and organ damage.
USP Chapter <61> testing is performed by plating the prepared sample onto two types of growth media. The sample preparation details are determined during method suitability. The growth medias and sample mixtures are incubated at defined temperatures and durations. After the incubation period, the colony numbers are counted, and the results are calculated to correct for any dilution of the compounded preparation that occurred during test-sample preparation. The test results are then compared to the defined acceptance criteria to determine if the nonsterile preparation passes or fails the test. The goal of USP Chapter <61> testing is to ensure that there is low bioburden in nonsterile compounded preparations and intermediates.
United States Pharmacopeia Chapter <62> Tests For Specified Organisms
While USP Chapter <61> demonstrates the total number of microorganisms, USP Chapter <62> tests for the presence of specific organisms. The FDA refers to this in 483s as tests for “objectionable organisms” or lists “specific organisms” that cannot be present in nonsterile preparations. For example, the FDA has stated “freedom from Pseudomonas aeruginosa” or “absence of bile-tolerant Gram-negative bacteria” when referring to lack of Chapter <62> testing. To determine which organisms listed in USP Chapter <62> should not be present in nonsterile preparations, you should consider the route of administration and USP Chapter <1111>. These tests are performed similarly to USP Chapter <61> but utilize growth media designed to promote the growth of the specific target microorganism and inhibit the growth of others. Method suitability is required to determine the appropriate sample preparation steps necessary to overcome any antimicrobial properties of the compounded preparation. This ensures that the test will detect the objectionable microorganism if present. At the conclusion of the incubation, a result of “pass” or “fail” is generated. A passing result means that the target organism was not detected in the sample. A failing result means the objectionable microorganism was found. The goal of USP Chapter <62> testing is to ensure that nonsterile compounded preparations do not contain specific microorganisms of concern.
Click Here to Access the Common Pharmaceutical Preservatives
United States Pharmacopeia Chapter <51> Antimicrobial Effectiveness Testing
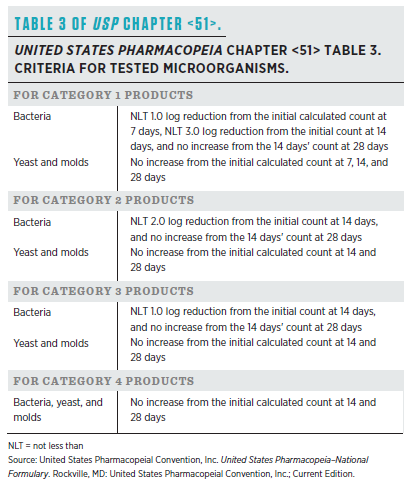
Many nonsterile and sterile compounded preparations contain antimicrobial preservatives to prevent the growth of microbial contamination throughout the BUD. The preservatives are needed because end users may introduce microorganisms into their compounded medication. Table 1 of this article lists common pharmaceutical preservatives, the type of formulation that is used, the effective concentration, optimal pH, and the types of organism the preservative is effective against. Examples of when microorganisms can be introduced into the compounded preparation are 1) when a patient puts unclean hands into their topical cream or 2) by repeated use of an oral product. Entry into a vial without proper aseptic technique can introduce microorganisms into sterile preparations. The test procedure described in USP Chapter <51> simulates these contamination events by adding high concentrations of 5 microorganisms to the compounded preparation. The effectiveness of the antimicrobial preservative is determined by counting the number of microorganisms present for 28 days following the contamination event. The counts occur similarly to USP Chapter <61> where the test sample is prepared, inoculated onto growth media, incubated, microorganisms counted, and results calculated to correct for any dilution of the compounded preparation while making the sample for counts. The sample preparation procedure for counting is determined during method suitability. The acceptance criteria for USP Chapter <51> vary based on the type of product and the type of organism. Table 1 in USP Chapter <51> divides the types of product into 4 categories based on the type of product and route of administration. Table 3 of USP Chapter <51> provides the acceptance criteria for each product category. Notice that each product category has 2 acceptance criteria. One is for bacteria, and the other is for yeast and molds. The goal of USP Chapter <51> testing is to prove that antimicrobial preservatives kill introduced microorganisms or prevent their proliferation.

United States Pharmacopeia Chapter <1207> Package Integrity Evaluation – Sterile Products
USP Chapter <1207> describes the tests that are used to demonstrate that container-closure systems can maintain a sterile environment throughout the BUD. The FDA says testing container-closure integrity is more likely to detect problems than sterility testing throughout the storage period. USP Chapter <1207> also discusses the potential for drug product components to escape from the container-closure system and the product quality risks posed by leaks. Escaping drug product components can be problematic for both sterile and nonsterile compounded preparations. For example, if benzyl alcohol in a preserved preparation escapes from the packaging system, the concentration may fall below its effective concentration range. Other components, such as water, could also escape, increasing the compounded preparation’s drug potency. Evaluating container-closure integrity is, therefore, important for both sterile and nonsterile compounded preparations. Many container-closure integrity tests are described in Chapter <1207>. The goal of USP <1207> testing is to ensure that the product’s package provides a level of protection required to meet physicochemical label-claim specifications and maintain product sterility until time of use. All the tests are designed to challenge the entire packaging system, not the individual components. Individual components are qualified by tests described in other USP chapters. USP Chapter <1207> divides the various tests into two categories: 1) deterministic and 2) probabilistic.
Deterministic Tests
Deterministic methods are described as quantitative and definitive and preferred by the USP. Vacuum decay is a common deterministic method. Vacuum decay is performed by placing the entire packaging system into a test chamber and drawing a vacuum on the chamber. The instrument then monitors for any changes in the vacuum level pulled on the chamber. Vacuum levels are monitored by the test instrument. Vacuum level changes indicate a leak in the packaging system. Because the test results are obtained from the instrument and not by an interpretation from the test operator, the USP prefers deterministic container-closure test methods.
Probabilistic Tests
Probabilistic methods are those that are qualitative and use human judgement. The USP considers these tests less desirable than deterministic methods. Dye ingress is a common probabilistic method. A common way to perform dye ingress testing is by submerging the container-closure system inside a chamber. The chamber is then filled with a dye and a vacuum pulled. The vacuum is then turned off and the chamber allowed to return to atmospheric pressures. Most of the time, the test articles are then visually examined for the presence of dye. The differences in color perception from one analyst to the next add variability to the test results. To overcome this variability, the drug product can be tested using an analytical instrument to test for the presence of dye. However, some drug products can oxidize the dye so even a high-performance liquid chromatogram with mass spectrometry would not detect the dye. Testing for container-closure integrity is important to determine if there is a leak in the packaging system that allows microorganisms to enter or if drug preparation components can escape. The USP considers probabilistic test methods, such as dye ingress, less desirable than deterministic test methods because analyst interpretation is often required to obtain the test results.
Conclusion
The USP has many resources for compounding pharmacists to ensure their patients receive quality compounded preparations. Compounders understand that USP Chapter <795> and USP Chapter <797> provide guidance on how to make quality preparations. Both chapters state that the compounding staff must be knowledgeable of the standards in USP Chapter <795> and/or USP Chapter <797> and must also be familiar with USP Chapter <1163>. This chapter provides information on QA and QC responsibilities within a compounding pharmacy. Part of QC’s responsibility is to execute the testing program setup by the QA group. USP Chapter <1163> also provides recommendations on which tests to perform on different types of compounded preparations. Compounding is heavily regulated so compounding personnel should utilize as many tools as possible to educate themselves. Testing itself is not intended to be the QA/QC program. Testing is a lagging indicator that all the things QA is doing are working appropriately. Without testing, patient complaint or harm may be the only feedback.
Resources
- United States Pharmacopeial Convention, Inc. United States Pharmacopeia–National Formulary. USP Chapter <795>; USP Chapter <797>; USP Chapter <1163>; USP Chapter <61>; USP Chapter <62>; USP Chapter <1111>; USP Chapter <51>; USP Chapter <1207>. Rockville, MD: United States Pharmacopeial Convention, Inc.; Current Edition.
- U.S. Food and Drug Administration. Guidance Document: Container and Closure System Integrity Testing in Lieu of Sterility Testing as a Component of the Stability Protocol for Sterile Products Guidance for Industry. [FDA Website.] February 2008. Available at: www.fda.gov/regulatory-information/search-fda-guidance-documents/ contaIner-and-closure-system-integrity-testing-lieu-sterility-testing-component-stability-protocol. Accessed January 20, 2021.
- Vu N, Nguyen K, Kupiec TC. Quality-control analytical methods: The essentials of United States Pharmacopeia Chapter <51> Antimicrobial Effectiveness Testing and its application in pharmaceutical compounding. IJPC. 2014; 18(2): 123–130.
- Vu N, Lou JR, Kupiec TC. Quality control analytical methods: Microbial limit tests for nonsterile pharmaceuticals, Part 1. IJPC. 2014; 18(3): 213–221.
- Vu N, Lou JR, Kupiec TC. Quality control: Microbial limits tests for nonsterile pharmaceuticals, Part 2. IJPC. 2014; 18(4); 305–310.