Abstract:
Potency tests, known as quantitative tests, are designed to determine how much of the active drug is in the sample. Stability tests are used to determine a beyond-use date for a preparation. Employing the proper method to determine potency or stability is key to understanding the difference between potency testing and stability testing. Methods of determining potency may or may not be stability indicating, but stability can be determined only by a stability-indicating method. A stability-indicating method can determine both potency and stability. Quality assurance programs are essential to establishing standards for compounded preparations. It is important that compounding pharmacists understand the differences between potency and stability tests and that these tests are made an integral part of the quality assurance program.
____________________________________________________________________________________
Oftentimes the question is asked “What is the difference between potency and stability?” This seems like a rather simple question and in some respects it is. To answer this question, however, the differences in the methods used to analyze the potency and stability of a compound must be understood. The most common mistake in determining stability is failure to use an analytical method that has been demonstrated to be stability indicating.1 It is not a surprise, therefore, that the most important aspect of determining potency and stability is the methods employed in the process. Simply put, a stability-indicating method must be used to determine stability. A stability-indicating method also can determine potency, but not all potency tests can determine stability.
The purpose of this article is to explain the difference between potency and stability, why they are important, and how they are determined. The method used to determine the concentration of the active ingredient, or analyte, is the most critical step in the process.
Quality Assurance in Compounding
United States Pharmacopeia (USP) Chapter <1075> Good Compounding Practices defines compounding as “the preparation, mixing, and assembling, packaging, and labeling of a drug or device in accordance with a licensed practitioner’s prescription of medication or under an initiative based on the practitioner/patient/pharmacist/compounder relationship in the course of professional practice.”2 The art of pharmaceutical compounding has long been a fundamental element in the profession of pharmacy.3 In an effort to ensure that each preparation is made appropriately and safely, a set of standards has been developed by the United States Pharmacopeial Convention, Inc. (USP). One such standard, included in Chapter <1075>, is the requirement that a compounded preparation be assigned a beyond-use date.2 The beyond-use date must be based on published data, appropriate testing (i.e., stability-indicating method), or USP–National Formulary standards.2 It is the compounding pharmacist’s responsibility to follow the USP guidelines when preparing compounded medications. Chapter <1163> Quality Assurance in Pharmaceutical Compounding defines a quality assurance program as “a system of steps and actions taken to ensure the maintenance of proper standards in compounded preparations.”4 A quality assurance program is essential to ensuring that the USP guidelines are met and that each compounded preparation is safe.
When compounding a new preparation that has not been tested appropriately, it is important that potency and stability studies are performed to determine concentration and a beyond-use date, respectively. A medication that is very potent (e.g., fentanyl) or has a narrow therapeutic index (e.g., levothyroxine) may produce a magnified (or greatly reduced) effect if its concentration is altered even slightly. Nitroglycerin, an antianginal medication that patients rely on to avoid chest pain, has a questionable stability profile. In the event that the beyond-use date is not accurate, and the patient does not experience relief because the medication is inactive, the consequences could be severe, including hospitalization and even death. Although these drugs may or may not be compounded, they simply serve as examples of the importance of determining potency and stability. In these situations, a small error can cause significant harm to the patient. It is imperative to understand the difference between potency and stability testing so that a concerted effort can be made to meet quality standards.
Potency
Potency is defined as the concentration of the drug in a compounded preparation.5 Potency tests are known as quantitative tests and are designed to determine how much of the drug is in the sample.3 High-performance liquid chromatography (HPLC) is the method typically employed in determining potency.5 HPLC is preferred because it is very specific and efficient. Although HPLC can be used in stability-indicating methods, not all HPLC procedures are stability indicating, and they must not be assumed to be so.6 Other methods used to test potency include titration, which uses the principles of chemistry, and microbial assays, which are sometimes used to test antibiotics.5 Titration is based upon a known chemical reaction with the tested drug.5 A microbial assay uses bacteria to examine what is known as “zones of inhibition” by the antibiotic in question.5 When used alone (without chromatography), ultraviolet (UV)/visible spectrophotometry can be employed to determine potency of a single analyte in solution. In this test, multiple compounds could interfere with absorption, yielding erroneous results. When performing a potency test, the method used determines whether stability also can be determined. As already mentioned, only a stability-indicating method can be used to determine stability.
This is where some compounding pharmacists run into problems. For example, let’s say your pharmacy contracts an analytical laboratory to run a potency test on your compound and you want results at day 0, 30 days, and 60 days. The target concentration of your compound is 10 mg/mL. The test performed indicates potency only, not stability. In other words, at the predefined time points of day 0, 30 days, and 60 days, the lab analyzes only how much of the compound is present. The method used could not differentiate the compound of interest from degradants or excipients in the preparation. The results reported indicate that the concentration of the compounded preparation was 10 mg/mL at each time point. This result cannot be extrapolated as representing stability at 60 days because the preparation may have contained degradants or excipients that were present but not detected by this analysis. To put it into numbers, for example, the reported concentration of 10 mg/mL may have comprised only 6 mg/mL of the active ingredient, while 3 mg/mL was degradants and 1 mg/mL was excipients. The most important thing to recognize is that some tests determine potency but not stability. Had stability-indicating methods been used to determine potency in this case, then the results could have been used to determine a beyond-use date (i.e., stability). If a stability-indicating method showed the concentration at time 60 days to be 10 mg/mL, you could be sure that the entire 10 mg/mL was active ingredient.
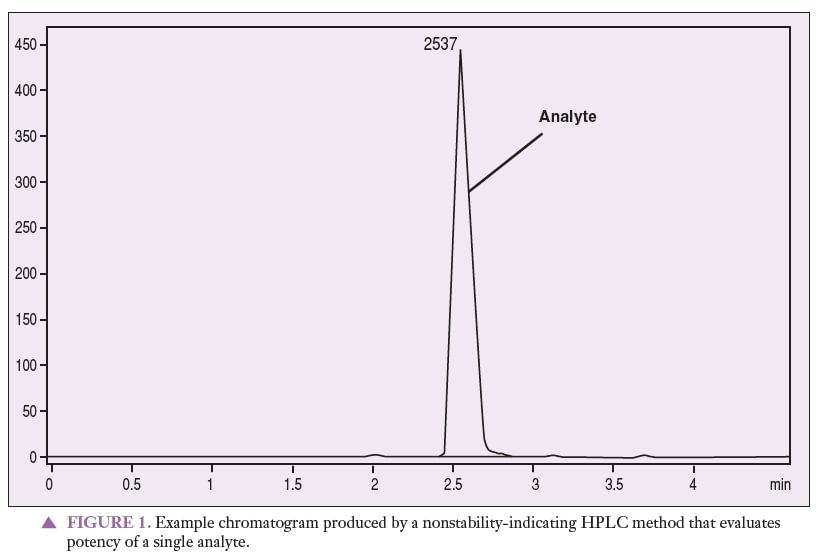
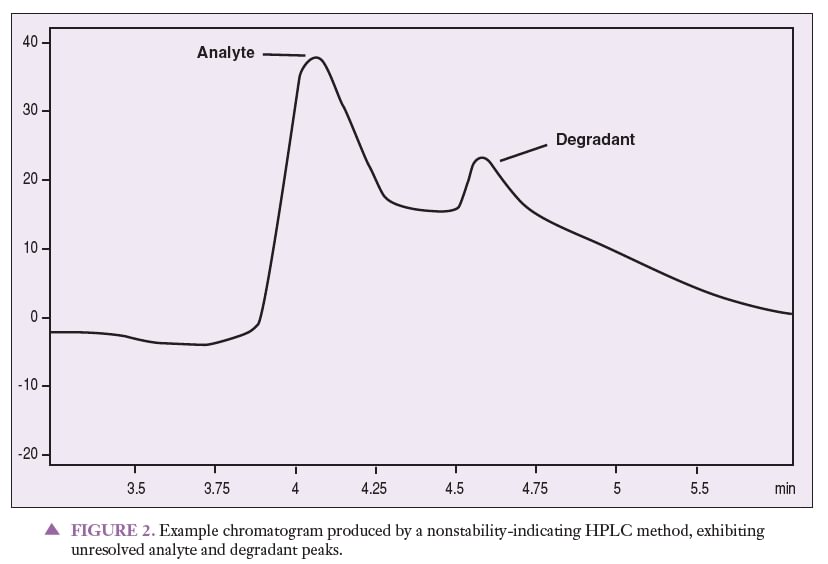
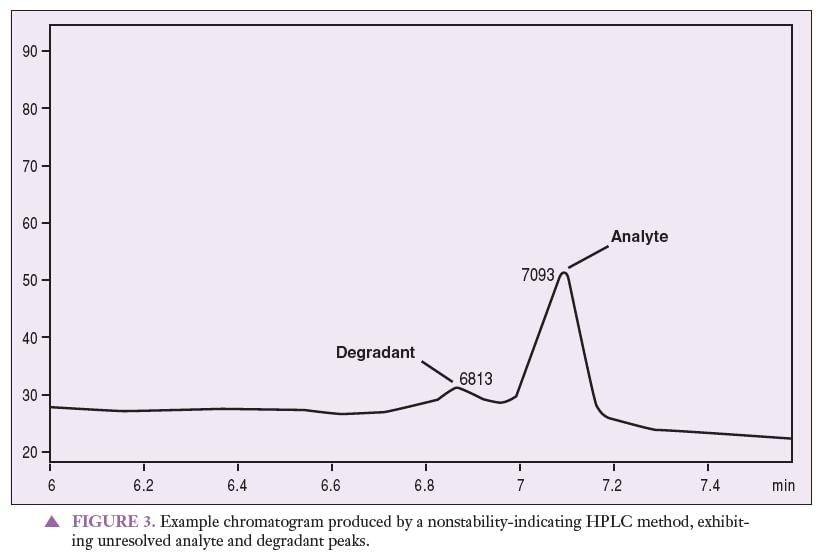
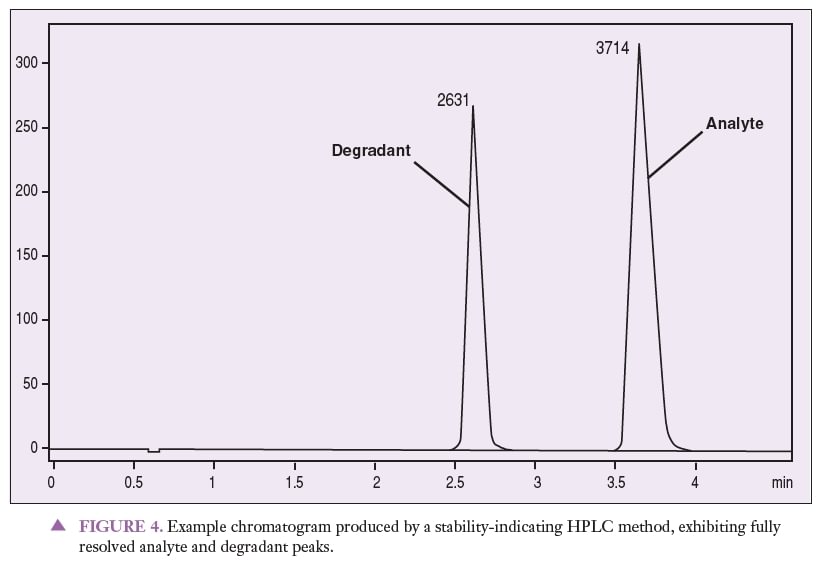
Figure 1 represents a chromatogram produced by a nonstability-indicating HPLC method that can be used to quantitate an analyte of interest. Figures 2 and 3 represent chromatograms produced by a nonstability-indicating HPLC method, exhibiting analyte and degradant sample peaks that are not resolved. All that can be concluded is that degradants were present in the sample at the time of the analysis. In Figures 2 and 3, no conclusions can be made about potency or stability. The peaks are not resolved, and thus it is impossible to properly quantitate the analyte (i.e., determine potency). Stability cannot be determined simply because stability-indicating methods were not used.
Stability
In USP Chapter <795>, stability is defined as “the extent to which a preparation retains, within specified limits, and throughout its period of storage and use, the same properties and characteristics that it possessed at the time of compounding.”2 This chapter defines beyond-use date as “the date after which a compounded preparation is not to be used and is determined from the date the preparation is compounded.”2 Stability testing is used to determine a beyond-use date, which is required by USP guidelines to be on the label or package of a compounded preparation.2 The terms stability, shelf life, and beyond-use date can be used interchangeably when referring to compounded preparations. The term expiration date is used when referring to manufactured products.
Chapter <797> Pharmaceutical Com- pounding—Sterile Preparations states, “It should be recognized that the truly valid evidence of stability for predicting beyond- use dating can be obtained only through product-specific experimental studies.”7 It is important to remember that the analytical method employed is key to determining stability versus potency. Once again, a stability-indicating method must be used to establish stability. Furthermore, a potency test that used stability-indicating methods can be used to determine stability as well as potency.
Stability testing usually includes method development, method validation, and a stability study. The method must separate the active ingredient from its degradants and impurities, as well as any other excipients in the preparation. This is done by force degrading the active ingredient and inactive ingredients to ensure that no degradants interfere with the analysis. In the process of force degradation, the compound is exposed to high heat and humidity, UV radiation, an acid, a base, and peroxide.8 It is this step that differentiates a stability-indicating test from a simple potency test. Figure 4 is an example chromatogram produced by a stability-indicating HPLC method, showing analyte and degradant peaks that are fully resolved from one another. When looking at this chromatogram, it is important to notice that the active ingredient is completely separated from its degradants. Stability can be determined from this type of study, simply because stability-indicating methods were used in the analysis.
Method validation ensures that the method meets certain criteria. The typical analytical characteristics used in method validation include accuracy, precision, specificity, detection limit, quantitation limit, linearity, range, and ruggedness, as outlined in USP Chapter <1163>. The stability study includes storing the preparation in stability chambers, testing it at predetermined time points, and then determining stability. These time points may be specified by the compounder or dictated by the particular compound. Once again, it is crucial to understand that the methods used to deter- mine stability must be stability indicating.
Quality assurance programs are essential to establishing appropriate standards for compounded preparations. The specific program implemented is up to the com- pounding pharmacy but should include a standard operating procedure, documentation, verification, and testing as outlined in USP Chapter <1163>.4 The standards of the Pharmacy Compounding Accreditation Board state that “a pharmacy must provide documentation of the basis for its determination of the beyond-use date assigned to its compounded preparation.”9
Conclusion
Employing the proper method to determine potency or stability is the key to understanding the difference between potency testing and stability testing. Methods of determining potency may or may not be stability indicating, whereas methods used to determine stability must be stability indicating. Stability-indicating methods can determine both potency and stability. It is easy to see that the madness truly lies in the methods.
References
- Trissel LA. Avoiding common flaws in stability and compatibility studies of injectable drugs.
Am J Hosp Pharm 1983; 40(7): 1159–1160.
- United States Pharmacopeial Convention, Inc. United States Pharmacopeia 27–National Formulary 25. Rockville, MD: US Pharmacopeial Convention, Inc.; 2007; 2 Supplement: 334, 511.
- Kupiec T, Huerta PL Jr. Analytical testing of extemporaneously compounded preparations. IJPC
2000; 4(2): 105–107.
- Kupiec TC. Quality-control analytical methods: Chemical testing aspects of USP Chapter <797> for compounded sterile preparations. IJPC 2005; 9(2): 136–138.
- United States Pharmacopeial Convention, Inc. United States Pharmacopeia 31–National Formulary 26. Rockville, MD: US Pharmacopeial Convention, Inc.; 2007. [In press.]
- Trissel LA. Stability studies: Five years later. Am J Hosp Pharm 1988; 45(7): 1570.
- [No author listed.] Chapter <797> Pharmaceutical Compounding—Sterile Preparations. PF 2007; 32(3): 952.
- Kupiec TC. Dr. Kupiec’s corner: What’s in a name? Beyond use dating–101, 201,301. The Pharmacists Link. Oklahoma City, OK: Analytical Research Laboratories, Inc.; Spring, Summer, Fall 2003.
- [No author listed.] Pharmacy Compounding Accreditation Board. PCAB standards with compliance indicators: Standard 6.10 beyond-use date. 2006; 7.2: 20.
Kupiec T, Skinner, R, Lanier, L. “Quality Control Analytical Methods – Stability Versus Potency Testing – The Madness is in the Method”. IJPC 2008; 12(1): 50–53
Request a Quote