
Kayla Lipcaman, ARL Bio Pharma Associate Microbiology Supervisor
Compounding pharmacies and outsourcing facilities must ensure that sterile compounded preparations are free from microbial contamination. To achieve this, they use aseptic processing. According to the United States Pharmacopeia (USP), aseptic processing is a method by which separate components (e.g., drugs, containers, or closures) are brought together under conditions that prevent microbial contamination.
A media fill, also known as a process simulation, is performed to evaluate the effectiveness of the aseptic process and the personnel’s aseptic technique in preventing contamination.
The Food and Drug Administration (FDA) has established guidelines for performing media fills to validate aseptic operations. Media fills should be conducted during the initial qualification and at least twice a year, or as required, to maintain aseptic process control. Additionally, if there are significant changes to the facility, equipment, processes, or test methods, a new media fill must be performed to re-validate aseptic processing.
USP has also established standards for performing media fills as part of a competency test to assess personnel’s aseptic technique. A media fill is required before beginning to compound Category 1, 2, or 3 CSPs, and at predetermined intervals for each drug product category.
Media Fill Design
During a media fill, a microbiological growth medium that has previously or concurrently passed growth promotion testing is used in place of a drug product. The growth medium is exposed to the same contact surfaces and process conditions encountered during routine aseptic production, including:
- Equipment
- Container closure systems
- Critical environments
- Aseptic process manipulations
Media fills must closely simulate routine aseptic operations and be performed under worst-case, most challenging conditions, as required by USP and FDA guidance. The simulation should reflect actual production practices and batch sizes. Critical factors to consider include:
- Line speed
- Batch size
- Number and qualifications of personnel
- Routine and non-routine operational practices
Growth Media
The most common growth medium for media fills is Soybean Casein Digest Medium (SCDM), also known as Trypticase Soy Broth (TSB). Growth media may be obtained from a qualified commercial supplier or prepared in-house:
- If using a commercial supplier, a certificate of analysis (COA) must be obtained, according to the FDA and USP, to confirm that the media lot supports microbial growth. 503A pharmacies and 503B outsourcing facilities are required to qualify their suppliers.
- If preparing growth media in-house, each batch must undergo documented growth promotion testing in accordance with USP 71 Sterility Tests, prior to use in a media fill.
Incubation and Observation
After completion of the aseptic process simulation using growth media, the final sealed containers are incubated and observed under controlled conditions to detect potential microbial contamination.
The FDA recommends that incubation may be performed at:
- A single temperature range of either 20–25°C or 30–35°C; or,
- A two-stage incubation approach may be used, consisting of incubation at 20–25°C for a minimum of 7 days, followed by incubation at 30–35°C for an additional minimum of 7 days.
USP recommends incubation at 20–25°C for a minimum of 7 days, followed by incubation at 30–35°C for an additional minimum of 7 days.
Interpreting Test Results
If microbial growth is observed during or after the incubation period, the contaminated unit must be investigated. A microbial identification may be performed, as appropriate, to identify the organism to the genus and species levels and to support the investigation.
Compounding pharmacies and outsourcing facilities should interpret results to assess the risk of a CSP unit becoming contaminated during actual operations (e.g., start-up, sterile ingredient additions, aseptic connections, filling, and closing).
According to the FDA, the recommended criteria for assessing the state of aseptic processing control are as follows:
- When filling fewer than 5,000 units, no contaminated units should be detected. One contaminated unit is considered a cause for revalidation, following an investigation.
- When filling 5,000 to 10,000 units, one contaminated unit should result in an investigation, including consideration of a repeat media fill. Two contaminated units are considered a cause for revalidation, following investigation.
- When filling more than 10,000 units, one contaminated unit should result in an investigation. Two contaminated units are considered a cause for revalidation, following an investigation.
According to USP, the recommended criteria for assessing the state of personnel competency in aseptic technique are as follows:
- No contaminated units should be detected.
- Media fill failure constitutes an overall failure of aseptic manipulation competency.
Contact ARL Bio Pharma today for more information on media fills at info@arlok.com or 800-393-1595.
Resources:
- USP 797 Pharmaceutical Compounding—Sterile Preparations
- USP 1116 Microbiological Control and Monitoring of Aseptic Processing Environments
- USP 1211 Sterility Assurance
- FDA Guidance for Industry – Current Good Manufacturing Practice – Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act
- FDA Guidance for Industry – Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice

Possible Causes and Investigation
A sterility test detects microbial contamination and provides data to determine whether a product is ready for release. A “Sterile” result indicates no contaminating microorganism is found in the sample. A “Not Sterile” result indicates microbial growth, and the product examined does not comply with the test for sterility, unless it is demonstrated that the test is invalid for causes unrelated to the product examined.
Sterility Test and Microbial Growth
Trained microbiologists examine the sterility test sample for microbial growth using the following conditions:
| Test Method | Criteria for Not Sterile Result |
|---|---|
| Traditional Sterility | Turbidity or opaqueness is observed during the incubation and subculture periods |
| Celsis Rapid Sterility (ATP Bioluminescence) | Relative light unit values exceed acceptance criteria |
| ScanRDI Rapid Sterility (Laser Scanning Cytometry) | “1 or more microorganism events” is detected and counted |
Once evidence of microbial growth is found, the test is out-of-specification (OOS), and an investigation is required to determine the cause of the test failure.
Possible Causes of Sterility Test Failure
- Compromised container integrity
- Improper aseptic processing and facilities
- Invalid sterilization processes
- Laboratory error
ARL Bio Pharma Sterility OOS Investigation Process
An OOS investigation is conducted for every sterility test failure. ARL Bio Pharma’s investigation includes a detailed examination of:
- Sterility test method
- Environmental monitoring (EM) data
- Sanitization logs for all ISO environments
- DNA sequencing reports, if applicable
- Deviation reports, if applicable
- Negative control data
- Potential retest results, if the test meets invalidation criteria
Criteria for Invalidating a Sterility Test Failure
A traditional sterility or Celsis rapid sterility test may be considered invalid only if one or more of the following conditions are fulfilled:
- A species-level microbial identification match between EM isolate(s) collected during the applicable testing shift and the microorganism isolate from the sterility test.
- Evidence that the testing procedure used during the test in question reveals a fault.
A ScanRDI rapid sterility test may be considered invalid only if both of the conditions are fulfilled.
If the test is declared invalid, it is repeated with the same lot and number of samples as in the original test. If no evidence of microbial growth is found in the repeat test, the product examined complies with the sterility test. If microbial growth is found in the repeat test, the product examined does not comply with the sterility test.
Next Steps
While a species-level match meets a portion of the USP 71 invalidation requirements, a sterility failure should prompt an investigation at the facility where the product was prepared. This investigation should take place at the same time as the laboratory investigation and include a review of:
- Compounding sterility assurance practices
- Aseptic processing practices
- Equipment qualifications
- Sterilization validations
- Environmental monitoring
- Personnel training
Pharmacies and outsourcing facilities should review all the data, including ARL’s findings, to determine how to proceed after a sterility test fails.
For more information, see USP chapters:
- USP 71 Sterility Tests
- USP 1207 Package Integrity Evaluation – Sterile Products
- USP 1211 Sterility Assurance
- USP 1229 Sterilization of Compendial Articles

Bailey Rubin, Technical Sales Representative
What happens to your compounded preparations after they leave your pharmacy? During transit, compounding preparations can experience shipping delays, which may result in temperature changes. Even the most carefully packaged preparations can be exposed to unexpected environmental changes. Without supporting data to demonstrate that a product remains stable under these conditions, a pharmacy could face compliance consequences, and patients could receive compromised medications.
Understanding Stability Studies and Temperature Excursions
Stability studies are required to demonstrate the stability of compounded preparations. They test how a preparation’s physical and chemical integrity performs under its intended storage condition(s) over time and provide data needed to support a Beyond-Use-Date (BUD). During the stability study, samples are stored in stability chambers that must be maintained within specifications that allow only very small changes in temperature and humidity. As stability samples are held under defined and controlled environmental conditions, stability studies are not intended to, nor can they, demonstrate the stability of compounded preparations under temperature excursions or conditions not included in the validated study parameters.
Temperature excursion studies are designed to evaluate how a compounded preparation responds when exposed to short-term temperature or humidity fluctuations outside its labeled storage range, such as 40°C ± 2°C / 75% RH ± 5% for 24 or 48 hours. After exposure to the higher temperature and humidity, the compounded preparations are then tested for stability using a stability-indicating method.
Why Temperature Excursion Studies Matter
Some compounds, such as GLP-1 (semaglutide, tirzepatide) and trimix formulations, can be highly susceptible to degradation when exposed to temperatures higher than those for which they are intended to be stored. Even just a few hours at room temperature or higher can lead to degradation, potentially compromising product quality. Regulatory authorities and quality standard organizations like the Food and Drug Administration (FDA), United States Pharmacopeia (USP), and International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) expect supporting data demonstrating that compounded preparations remain stable during a deviation from their labeled storage condition(s).
The FDA has repeatedly issued 483s and warning letters for failing to investigate temperature fluctuations. Examples of these observations include:
Firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed (21 CFR 211.192).
- “During the inspection, firm personnel indicated that this lot had experienced high temperature excursions while in storage.”
- “You had prolonged problems assuring maintenance of the required temperatures of the refrigerated storage unit in which this API was stored.”
Firm failed to establish a written testing program designed to assess the stability characteristics of drug products and determine appropriate storage conditions and expiration dates. Firm also failed to have buildings used in the manufacture, processing, packing, or holding of drug products with adequate space for the orderly placement of equipment and materials to prevent mix-ups and contamination (21 CFR 211.166(a) and 21 CFR 211.42(b)).
- Inadequate Material Storage – “Bulk totes of ethanol hand sanitizer gel were stored outdoors, and you informed our investigator that these totes had been stored outdoors since April 2020. Labeling for the bulk ethanol hand sanitizer gel lists a temperature storage range of (b)(4) F. The totes are exposed to temperature excursions that often exceed the specified temperature storage range, which could compromise the quality of the drug product therein.”
Firm failed to review and investigate production and QC laboratory deviations.
- “The inspection documented that, despite the fact that your firm has an uninterrupted power supply used by the QC laboratories, power failures have impacted the QC stability chambers. However, in each case, no investigation was conducted to determine the impact of the power loss on the samples kept within the chambers.”
Firm’s quality unit is not involved in quality related matters; the unit fails to review deviations from established specifications or procedures and does not adequately assess the need for corrective actions for deviations it is made aware of.
- “No follow-up or investigations were conducted for the excursions … to determine the root cause and potential impacts on the products and stability studies.”
Written procedures are lacking, which describe in sufficient detail the storage of components, drug product containers, and closures.
- “Specifically, there is no temperature monitoring in the warehouse where raw material and blended drug products are stored.”
Be Proactive
ARL Bio Pharma offers complete stability and temperature excursion testing that meets USP, FDA, and ICH standards. Our studies show how preparations hold up under different temperature and humidity conditions and provide the data needed to support beyond-use dates and quality claims.
Contact ARL Bio Pharma at 800-393-1595 or info@arlok.com today to see how a temperature excursion study can strengthen your stability program, protect patients, and keep your pharmacy compliant.
References:
- Food and Drug Administration
- ICH Stability Testing of New Drug Substances and Products Q1A(R2)
- USP 659 – Packaging and Storage Requirements
- USP 797 – Pharmaceutical Compounding – Sterile Preparations
- USP 1079 – Risks and Mitigation Strategies for the Storage and Transportation of Finished Drug Products
- USP 1149 – Guidelines for Assessing and Controlling the Physical Stability of Chemical and Biological Pharmaceutical Raw Materials, Intermediates and Dosage Forms

James Zellner, Technical Sales Representative
The FDA defines photostability as the ability of a drug substance or product to resist chemical changes when exposed to light and considers photostability testing as an “integral part of stress testing”. Much of the FDA’s standing on photostability and the associated studies to perform comes from the ICH Q1B Guidance for Industry Photostability Testing of New Drug Substances and Products. The FDA has officially adopted this document as guidance, so it should be used to design photostability studies.
ICH recommends a systematic approach to photostability testing, which, as appropriate, may include testing of the drug substance, exposed drug product (out of its primary packaging), drug product in any intermediate packaging, and, finally, drug product in its final packaging. Later, if there are changes to either the formulation or packaging, ICH states these studies should be repeated to demonstrate that no change to product stability occurs.
When conducting a photostability study, Q1B is specific in what types of light, wavelength, and exposure should be used. For visible light, this consists of not less than 1.2 million lux hours and a near UV exposure of 200-watt hours/ square meter. Exposure should be measured via chemical actinometric systems and/or calibrated radiometers/ lux meters. Throughout the exposure period, the temperature of the photostability chamber must be maintained to avoid additional stressors on the product that may lead to inaccurate photostability conclusions. Sample sizes should be adequate to demonstrate that no variability exists between individual product units. While there isn’t a specific number used in Q1B, 20 units are cited as an example for a solid dosage form. “Dark control” samples are included for comparison. During light exposures, the test samples are arranged in the chamber to ensure uniform exposure of the product to the light source and continue until the appropriate amount of lux/ watt hours is achieved.
At the conclusion of the exposure period, many tests may need to be performed to assess photostability. The drug substance or product should be assayed for stability of the API (active pharmaceutical ingredient) using a formulation-specific, stability-indicating analytical method. As with other guidance, FDA expects this to be done via a chromatographic method that is reliable, meaningful, uses a reference standard, and is highly specific. The assay method should have, during its development, been created using both visible light and UV stressors to ensure the potential degradants created during this exposure are separated from the API. Testing to assess photostability should not be limited to the assay. Appearance, color and clarity of solution (in the case of a liquid product), and dissolution/disintegration (in the case of a solid product) are examples of other tests that may need to be assessed to fully understand the photostability of the drug product. The appropriate tests to include depends on the drug substance or product being tested. Dark control samples are tested using the same scheme as the primary samples to compare.
Evaluation of the test results should include a determination of whether changes observed, if any, are acceptable. The results from stability studies should be considered when evaluating photostability data. If the substance or product is not stable when exposed to light, an assessment of the product or packaging systems is needed. Special labeling or packaging may be required to mitigate the effects of light exposure.
Contact ARL today to learn more about Photostability Studies and Testing. 800-393-1595 or info@arlok.com.

Gary Rhodes, Analytical Associate Laboratory Supervisor
Total Organic Carbon (TOC) testing measures the amount of carbon present in a water sample and is conducted based on USP 643 requirements. Chemical impurities can enter water from various sources, including:
- Source water used for pharmaceuticals (such as injectables)
- Packaging for sterile and non-sterile water
- Purification systems
- Processing equipment
This test serves as a key quality indicator, as organic carbon in water can promote microbial growth and signal potential contamination, posing risks to patient safety.
While a connection exists between TOC levels and microbial activity, a direct numerical correlation does not exist. Therefore, TOC measurements should not be used as a substitute for endotoxin or microbiological control testing.
TOC Test Method
ARL utilizes a Shimadzu TOC-LCSH, High-Sensitivity Analyzer to conduct the TOC test method. TOC is measured by injecting a volume of the sample into a combustion tube containing an oxidation catalyst heated to 680°C. The sample is burned in the tube, converting it into carbon dioxide, which is then measured by the non-dispersive infrared (NDIR) gas analyzer. This measurement is compared to a standard solution prepared from Sucrose at the specified limit to determine the amount of carbon in mg/L.
While this measurement quantifies the amount of carbon in the sample, it does not identify the specific contaminant. USP has established acceptance criteria for carbon content, ensuring that materials meeting these specifications are deemed suitable for their intended pharmaceutical application. If it does not meet requirements, further analysis and/or investigation is required to determine the source and identity of the contamination.
Definition and Limits
USP 643 sets limits for Bulk Water and Sterile Water. Bulk Water is defined as Purified Water, Water for Injections, Water for Hemodialysis, and the condensate of Pure Steam. The limit for carbon contained in samples of these waters is 0.5 mg/L or 500ppb of carbon.
Sterile Water is defined as Sterile Water for Injection, Sterile Purified Water, Sterile Water for Irrigation, Sterile Water for Inhalation, and any specific monograph that references Sterile Water. These waters are derived from Purified Water or Water for Injections; therefore, they have been determined to be compliant with the Bulk Water requirements before being stored and sterilized in their container. The limits for carbon contained in samples of these waters are dependent on the nominal container volume, as stated below.
USP 643 – Table 1. TOC Limit Based on Container Volume
| Nominal Container Volume (mL) | Limit 1 (L1) (mg/L of carbon) | Limit 2 (L2) (mg/L of carbon) |
|---|---|---|
| ≤5 | 32.00 | 48.00 |
| >5 and ≤100 | 24.00 | 36.00 |
| >100 | 8.00 | 12.00 |
Other Applications for TOC Testing
In addition to testing water, TOC is used for cleaning validations. It is suitable for testing direct surfaces and rinse water. However, TOC does not identify which compounds contain oxidizable carbon. Any carbon detected is attributed to the target compound(s) and compared to a set limit. If testing for a specific compound, contact ARL to discuss a residual drug cleaning validation study.
For more information on TOC, contact ARL Bio Pharma at 800-393-1595 or info@arlok.com.
Resources:
- USP643 Total Organic Carbon
- Shimadzu TOC-L Series Manual
- FDA Guidance for Q&A on Current Good Manufacturing Practice Requirements – Equipment
- Total Organic Carbon Testing – Back to Basics: American Pharmaceutical Review

The Food and Drug Administration (FDA) requires dissolution testing to ensure continuous drug product quality and performance of controlled-release and extended-release products. This testing is performed during product development, batch release, and stability studies to verify that the release rate of the active pharmaceutical ingredient meets established specifications. Variability in release rates between batches can pose a significant risk to patient safety and may result in inaccurate dosing, reduced drug efficacy, and adverse reactions.
Drug absorption is formulation-dependent and influenced by the active pharmaceutical ingredient’s release profile. The FDA uses a biopharmaceutics classification system (BCS) to categorize drug substances based on their aqueous solubility and intestinal permeability, which includes:
- Class 1: High Solubility – High Permeability Drugs
- Class 2: Low Solubility – High Permeability Drugs
- Class 3: High Solubility – Low Permeability Drugs
- Class 4: Low Solubility – Low Permeability Drugs
The BCS framework sets dissolution expectations and can support biowaivers for oral drug products. However, the FDA does not set a universal dissolution standard for each drug class. Instead, dissolution specifications are generally derived from United States Pharmacopeia (USP) monographs, when available, or developed and validated based on specific drug products. If there is no existing standard for a certain drug profile, compounders can establish their own specifications. Validation of the dissolution test method is required to support specifications and acceptance criteria. The dissolution characteristics should be based on the pH, solubility, and pharmacokinetics of the drug to ensure bioequivalence for future batches while also adhering to acceptable dissolution limits.
According to the Center for Drug Evaluation and Research, once a dissolution specification is set, the drug product should comply with that specification throughout its shelf life.
Dissolution Test Methods
Dissolution testing uses various apparatus methods based on USP 711. The basket and paddle methods are among the most commonly used for testing a wide range of drug products. In these methods, the solid dosage form is placed into a dissolution instrument containing six vessels, all maintained at a specific temperature. The paddle rotation speed and duration are controlled to simulate gastric or absorption conditions. At predetermined time intervals, samples are drawn from each vessel.
These filtered samples are then analyzed to quantify the percentage of active pharmaceutical ingredient (API) released from the dosage form, using UV spectrophotometry or HPLC. This allows for a precise assessment of drug release profiles.
Overall, dissolution testing is a critical component of quality testing programs. This testing provides valuable insights into the absorption rates, helps predict the release behavior of active pharmaceutical ingredients, guides the selection of the most effective formulations for optimal therapeutic outcomes, and ensures consistency throughout the production process.
For more information on dissolution testing, contact ARL Bio Pharma at 800-393-1595 or info@arlok.com
Resources:
- USP 711 Dissolution
- FDA Guidance for Industry Dissolution Testing and Acceptance Criteria for Immediate-Release Solid Oral Dosage Form Products Containing High Solubility Drug Substances
- FDA CDER Guidance for Industry Dissolution Testing of Immediate Release Solid Oral Dosage Forms

Potency testing measures the concentration of the active pharmaceutical ingredient (API) in a drug product. This critical test should be conducted before drug distribution, confirming that the API concentration is within specification of the labeled amount.
USP sets the acceptable potency range of ±10% for compounded preparations. However, this range may vary depending on the API, for example, ±20% for certain proteins and as narrow as ±5% for potent analgesics. Pharmacies may also establish internal specifications based on the specific formulation and its intended use.
When compounding with a pre-filled IV bag, the total volume of the bag varies based on the manufacturer’s excess volume, known as overfill, and the compounding method used. Pharmacies may:
- Add active pharmaceutical ingredient(s) to the solution; or,
- Withdraw solution before adding the active pharmaceutical ingredient(s) to the solution
ARL recommends adding a fill volume test with a potency test to ensure quality and consistency of potency results in the final preparation. The fill volume test measures the total volume of the IV bag, which includes the base solution volume, overfill, and the admixture volume.
After the total volume is determined, the laboratory uses this measurement to calculate potency results, reflecting the drug concentration per bag. Without this total volume assessment, potency results for IV bags may appear artificially low and out-of-specification (OOS), even if the correct drug amount was added.
Example: Two IV bags labeled as 100 mL contain different overfill amounts, such as 104 mL and 107 mL. If the pharmacy adds the same API quantity to both the 104 mL and 107 mL bags, the API concentration will differ between the two bags. This variation may lead to one bag’s potency results within specification, and the second bag’s results out of specification.
Pharmacies can minimize the risk of potency failures by ordering a fill volume test with a potency test for IV bags, ensuring that the drug concentration accurately reflects the API of the finished drug product. For more information on potency testing for IV bags, contact 800-393-1595 or info@arlok.com.
USP 797 Pharmaceutical Compounding – Sterile Preparations outlines the minimum standards for preparing compounded sterile preparations (CSPs) for human and animal medications. These standards are designed to ensure the quality of CSPs and to minimize potential harm to patients.
CSPs are distinguished into three categories (1, 2, and 3) based on the following criteria:
- Compounding environment in which the CSP is prepared
- Likelihood of microbial growth
- Time period for use (beyond use date)
Category 2 CSPs:
- Must be prepared in a cleanroom suite
- May be assigned a beyond use date (BUD) of >12 hours at controlled room temperature or >24 hours, if refrigerated, not to exceed the longest permitted BUDs in Table 13
- Requires stability data to support BUD
Personnel Qualification and Environmental Monitoring
Maintaining control of environmental conditions is crucial in a compounding pharmacy. USP requires all personnel who compound or supervise compounding personnel to complete initial competency assessments, followed by ongoing assessments to maintain quality assurance and quality control.
Click here to download ARL’s Handout on personnel qualification and environmental control requirements.
Release Inspections and Testing
Ensuring the quality characteristics of a sterile compounded preparation is essential for patient safety. USP requires documentation, visual inspections, and release testing to maintain sterility assurance and verify that the preparation meets quality standards before CSPs are released for patient use.
Click here to download ARL’s Handout on Category 2 Testing requirements.
| Release Inspections and Testing | Category 2 USP Testing Requirements |
|---|---|
| Antimicrobial Effectiveness Testing | For preservative aqueous multiple-dose CSPs, antimicrobial effectiveness testing must meet USP 51 requirements |
| Bacterial Endotoxin Testing | Required for injectable CSPs compounded from one or more nonsterile component(s) and assigned a BUD that requires sterility testing; and, Should be tested for injectable CSPs compounded from one or more nonsterile component(s) and assigned a BUD that does not require sterility testing |
| Container Closure Integrity Testing | Required for multidose containers and must pass the container-closure integrity test once for each formulation |
| Sterility Testing | Required for CSPs assigned a BUD that requires sterility testing |
| Visual Inspection | Must be performed at the completion of compounding and before release and dispensing |
ARL Bio Pharma supports your commitment to patient safety and meeting USP standards. Contact ARL at 800-393-1595 or info@arlok.com to learn more about our testing services and how we can assist with your quality testing requirements.
Antibiotics contain antimicrobial ingredients that either kill bacteria or prevent their growth. However, antibiotics can still pose risks to patients due to potential microbial contamination. It is crucial for pharmacies to ensure microbiological safety through a combination of process controls and sterility testing.
Sterility testing confirms the absence of microorganisms in a drug product. Because of their antimicrobial properties, antibiotics can interfere with sterility tests designed to detect microbial contamination. The extent of this interference may vary based on the active ingredients, inactive ingredients, preservatives, and the vehicle used in the formulation. No contaminating microorganisms can be present in the sample under the specified test conditions for a drug product to pass sterility testing.
Sterility test conditions include:
- Use of culture media that promotes microorganism growth
- Test methods selected based on sample type, container, volume, and filterable or non-filterable products
- Number of samples tested
- Incubation of media and examination for microbial growth
Method suitability is required for all formulations and test methods to demonstrate the validity of the sterility test. For the membrane filtration sterility test method, the preferred method according to USP 71, the sample is passed through a set of filters, the filters are rinsed, and each filter is placed in contact with growth media and incubated. In the direct inoculation sterility test method, the sample volume is inoculated into each growth medium and then incubated.
ARL Bio Pharma may add steps to the test method to ensure the effectiveness of the sterility test for antibiotics. These steps help eliminate or neutralize antimicrobial activity, making it possible to detect contamination, if present.
Additional steps for antibiotic sterility testing may include:
- Use of neutralizer or inactivating agents (e.g., Beta-lactamase to inactivate beta-lactam antibiotics)
- Utilizing a low-binding filter to ensure inhibitory residues are fully removed
- Pre-filtration dilution step before passing the sample through a filter
- Neutralizer added to the rinse medium
- Increasing rinse medium volume
The method is considered valid once the sample successfully passes method suitability testing by demonstrating that the inhibitory or antimicrobial properties do not prevent the sterility test’s ability to detect viable microorganisms. Alternatives to membrane filtration and direct inoculation test methods are acceptable, provided the method is validated in accordance with USP 1223 and method suitability is established.
Visit ARL’s Client Portal for antibiotic sterility testing sample requirements, pricing, and sample submission.
What is the difference between Potency and Stability?
It’s important to understand the purpose of each test to identify the difference between potency and stability.
Potency vs. Stability Testing
Potency testing verifies that the active pharmaceutical ingredient matches the identity and strength stated on the label. This essential quality check is used for formulation development, process and personnel qualification, and lot release. It is typically performed within a few days after compounding and provides a real-time assessment of the drug product’s quality.
Stability testing evaluates several factors of a compounded drug product’s formulation to determine a beyond-use date (BUD). It is typically performed after product development, and takes weeks to months to evaluate.
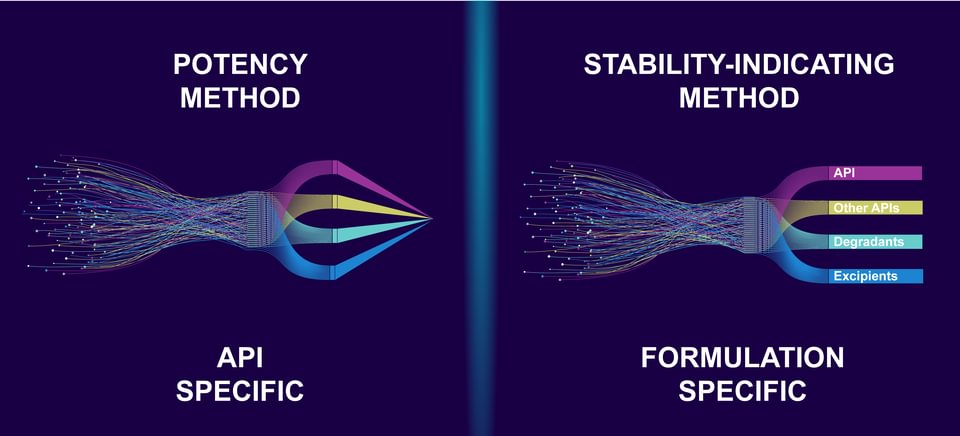
API Specific vs. Formulation Specific Methods
When ARL establishes a non-validated potency analytical method, the active pharmaceutical ingredient (API), not the specific formulation, is evaluated. The laboratory uses system suitability to verify that the instrument’s resolution and repeatability are adequate for analysis to provide reliable results. The analytical method is then used on API-specific lots for release testing. If an analytical challenge is encountered during potency testing for unique formulations, ARL will optimize the method and document changes to achieve accurate results.
When ARL develops a validated stability-indicating analytical method, the specific formulation is evaluated. At the beginning of method development, stress studies are conducted using acid and base, oxidation, heat, humidity, and light to quickly create degradants of the API and excipients that may appear during stability testing. Before it can be considered fully developed, the analytical method must demonstrate its ability to effectively separate the API from degradants, impurities, and excipients. Once the method is established, it must undergo validation for accuracy, linearity, precision, range, specificity, and system suitability. This validation is crucial so that the method can be used to assess the stability of the drug product and assign appropriate BUDs. Stability-indicating analytical methods may also be used on formulation-specific lots for release testing.
ARL Certificates of Analysis and Test Methods
If a drug product is tested by a non-validated potency analytical method, ARL’s certificate of analysis will have an instrument (HPLC) listed as the test method and a statement “the potency method(s) used for testing passed system suitability requirements per ARL SOP AMP-012 for non-GMP analysis. Product specific method validation is not available for the sample and specification(s) are for informational purposes only.”
If a drug product is tested by a validated stability-indicating analytical method, ARL’s certificate of analysis will list a method number (e.g., AMI-1234) as the test method, indicating the product was tested using the formulation-specific method.
For more information on potency and stability, visit ARL’s Education Center.