Andrew Taylor, Microbiology Supervisor
Microbial identification is an important component of a quality assurance program; specifically as it relates to environmental monitoring (EM), raw material and final product bioburden assessment, and out-of-specification (OOS) investigations.
Environmental Monitoring
USP <1116> Microbiological Control and Monitoring of Aseptic Processing Environments states “a successful environmental control program includes an appropriate level of identification of the flora obtained by sampling.” Continuous monitoring of the environment allows a facility to determine if any unusual microorganisms are present and is necessary for evaluating the effectiveness of the cleaning and sanitization measures. USP <1116> also speaks to the importance of analyzing contamination trends in any aseptic environment, which has long been a component of a successful environmental control program. Without microbial identification of EM isolates, it is very difficult to recognize when control over an environment has changed.
Another critical aspect associated with EM in a compounding facility is the monitoring for highly pathogenic microorganisms (e.g., Gram-negative rods, coagulase positive staphylococcus, molds and yeasts), which can be potentially fatal to patients receiving compounded sterile products. Should these organisms be found, USP <797> directs that it “shall be immediately remedied, regardless of cfu count, with the assistance of a competent microbiologist, infection control professional or industrial hygienist.”
Bioburden Assessment
It is important for a pharmacy to know of the specific microorganisms that may reside in their controlled environments and in their starting materials. It is becoming more common that pharmacies perform a bioburden assessment of their raw materials and non-sterile products as described in USP chapter’s <61> and <62>. When performing these tests, it is prudent to identify any contaminants found, and in the case of absence of specified organism testing, the need for microbial identification is specifically cited in USP <62>. This chapter requires confirmatory identification tests for any organisms that grow on selective media to determine a final result. Additionally, pharmacies should be aware if their raw materials contain a high bioburden to prevent potential out-of-specification results. For example, a downstream failure in endotoxin testing may be avoided if testing identifies gram-negative bacteria present in the raw material prior to formulation.
Out-of-Specification Investigations
According to USP <1116>, microbial identification can be useful data for investigating an out-of-specification result and looking for the possible contamination source. Once an organism’s identity is known, it becomes more practical to assess its possible ingress into an aseptic environment. For example, one may conclude that a soil-borne organism was likely to have been brought in on an employee’s shoes. This information can be part of the root cause assessment, where a conclusion may be that improper gowning techniques or insufficient contamination controls between outside shoes and an aseptic processing environment were to blame for an excursion or OOS event. It is also noteworthy that when performing USP <71> sterility testing, the general test chapter allows for invalidation of an OOS result if identification of the microorganisms isolated from the test may be “unequivocally ascribed to faults with respect to the material and/or the technique used in conducting the sterility test procedure.”
Microbial identification helps improve the quality of pharmacy operations and provides an increased understanding of compounding environments.
For more information, contact ARL at 800-393-1595 or info@arlok.com.
References
– USP <61> Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests
– USP <62> Microbiological Examination of Nonsterile Products: Tests for Specified Microorganisms
– USP <71> Sterility Tests
– USP <797> Pharmaceutical Compounding—Sterile Preparations
– USP <1116> Microbiological Control and Monitoring of Aseptic Processing Environments
Andrew Taylor, ARL Bio Pharma Microbiology Supervisor
In recent years, there have been instances of endotoxin-caused illness resulting from contaminated compounded sterile preparations. In 2015, an incident occurred where seven cases of endotoxin poisoning were related to contaminated glutathione infusions. In this case, patients were symptomatic within 2 hours of administration. Most cases reported fever, rigor, and headache. An investigation into the compounding pharmacy identified multiple issues with aseptic production of compounded products, including minimal compliance with standard quality assurance guidelines. Further testing of the glutathione powder itself revealed high levels of endotoxins.
Bacterial endotoxins are remnants of bacterial cells and are not detected by a sterility test. Potential sources of endotoxins include water, packaging components, equipment, and chemical / raw materials used during compounding of a drug product. Each drug has an allowable endotoxin limit, which specifies the amount of endotoxin that can safely be present. Criteria for acceptance is based on human tolerance and generally defined by the USP monograph, or by a calculation using the patient’s weight, route of administration, and maximum bolus dose. Some firms will also include a “safety factor” in their calculation to deliver a more conservative limit for a product.
It’s important that aseptic production of compounded products include in-process checks to monitor the presence of bacterial endotoxins. The FDA describes expectations in more detail by stating that “firms should take into account aspects of the manufacturing design, including consistency of a manufacturing process, impact of in-process hold times, endotoxin removal steps, and finished product endotoxin specifications” as they design their sampling plans. The plans should be dynamic; in that it may be necessary to adjust sampling when a deviation or an in-process change occurs. It’s also important to define an effective depyrogenation process as endotoxins are not removed by filter or steam sterilization.
USP <797> requires a Bacterial Endotoxin Test (BET) for each preparation of:
- Category 2 and Category 3 CSPs compounded from one or more nonsterile component(s)
- Multiple-dose CSPs
USP <797> also requires a description of the depyrogenation process employed, including the temperature, pressure (if applicable), duration, permissible load conditions for each cycle; and the use of endotoxin challenge vials (ECVs) must be included in the facility’s SOPs. The chapter also states that if a CSP is dispensed or administered before Endotoxin testing results are known, a facility must have procedures in place to Immediately notify the prescriber of a test failure with the potential to cause patient harm.
The Food and Drug Administration (FDA) requires a BET for all 503B outsourcing facility drug products reported to be non-pyrogenic.
Even if it is not mentioned specifically in the regulatory documents, it is important to check for the presence of endotoxins in raw materials, at various points in the compounding process, and in finished products before administering a drug to the patient. Good quality practice requires control or monitoring of the endotoxin levels of contamination at all steps of the compounding process. This should also include a thorough investigation if bacterial endotoxin contamination is found. Appropriately monitoring the presence of bacterial endotoxin in a compounding pharmaceutical environment is essential for your patients’ safety.
For more information on endotoxin testing, contact ARL at 800-393-1595 or info@arlok.com.
References
United States Pharmacopeia <85>
United States Pharmacopeia <797>
Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers. U.S. Department of Health and Human Services Food and Drug Administration. June 2012.
Johnstone, T et al. “Seven cases of probable endotoxin poisoning related to contaminated glutathione infusions.” Epidemiology and infection vol. 146,7 (2018): 931-934. doi:10.1017/S0950268818000420
Kerri Hirst, Microbiologist, ARL Bio Pharma
A sterility test detects microbial contamination and provides data to determine if your product is ready for release. A “Sterile” result indicates no contaminating microorganism is found in the sample. A “Not Sterile” result indicates microbial growth and the product examined does not comply with the test for sterility, unless it is demonstrated that the test is invalid. An investigation is performed to determine if a sterility failure is invalid due to laboratory error.
Sterility Test Failure (Out of Specification)
Once a sterility test is initiated, trained microbiologists periodically observe growth media for turbidity or fogginess indicating microbial growth. Microbial contamination may appear at any point during the incubation (Day 1-14) and subculture (Day 14-18) period. Once contamination is confirmed, the test is out-of-specification (OOS) and an investigation is required to determine the cause of a failed sterility test.
Possible Causes of Sterility Test Failure
- compromised container integrity
- improper aseptic processing and facilities
- invalid sterilization processes
- false positives
For more information on potential causes see USP chapters:
- USP <71> Sterility Tests
- USP <1207> Package Integrity Evaluation – Sterile Products
- USP <1211> Sterility Assurance
- USP <1229> Sterilization of Compendial Articles
ARL Bio Pharma’s Sterility OOS Investigation Process
An OOS investigation is conducted for every sterility test failure. This investigation consists of:
- detailed examination of sterility test method
- environmental monitoring data
- sanitization logs for ISO 8, 7 and 5 environments
- DNA sequencing reports
- deviation reports (if applicable)
- negative control data
- potential retest results, if the test meets invalidation criteria
ARL Bio Pharma’s criteria for invalidating a sterility failure is a species level microbial identification match between ISO 5 environmental monitoring isolate(s) from the days surrounding the test date, or evidence that the test was compromised due to sterility test controls or materials failures. If there is a microbial identification match or failure of the test controls/materials, a sterility retest using the same lot, number of articles, and test method completes the criteria for invalidation.
A match between an organism from the sample and ARL’s environment does not guarantee that contamination occurred during sterility testing, as common organisms may also be present in the original compounding environment. While a species level match satisfies a portion of the invalidation requirements described in USP <71>, a sterility failure should prompt an investigation of compounding sterility assurance practices, equipment, and environmental monitoring at the facility in which the product was prepared.
ARL has observed sterility failures due to multiple causes. In one instance, a “not sterile” result was invalidated due to a cracked filter container. The growth media used to test a Vancomycin product for sterility was turbid at the 14 day observation point, and, upon further examination of the media container, a crack was discovered. A discussion with the media container manufacturer confirmed this was a likely source of contamination. A retest using the same number of articles was performed, using the same test method, and a sterile result was produced which invalidated the original OOS result.
While ARL investigates each sterility failure thoroughly, it does not have access to client data regarding sterility assurance practices, equipment qualification, sterilization validations, personnel training or environmental monitoring. The only information ARL can utilize in investigating the cause of a sterility failure is internal data, which allows for determination of false positives or failure of test controls/materials. Any other sterility failure causes can be determined using data from the facility in which the product was compounded. Pharmacies and outsourcing facilities should assess all data to determine how to proceed following a sterility test failure.
Importance of Forced Degradation in Stability-Indicating Methods
Pharmacists often want to assign a beyond-use date (BUD) on a drug product longer than USP guidelines to increase compounding efficiencies and reduce drug waste. For accurate BUD assignment, Stability-indicating methods are recommended as opposed to Potency methods. Stability-indicating methods are specifically designed to separate drug degradants from non-degraded drug (for more information about the difference between the two methods, see Stability vs. Potency testing article).
Forced degradation uses stress conditions to create degradants in a drug product. This is required to demonstrate specificity in a stability indicating method and ensure an active ingredient’s chemical stability over time.
Quantification of analytes
Potency and stability are generally measured by a (U)HPLC coupled with a detector (overview of HPLC technology). These methods work in two steps. First, the method must separate the ingredients in the drug product, and then measure a signal for detected compounds. The result is translated into a chromatogram (Figure 1), a plot that gives the signal measured over time. If the method is well developed, each peak on the chromatogram corresponds to one compound. In this figure the test method is stability indicating because it separates Edetate Disodium from its degradant.
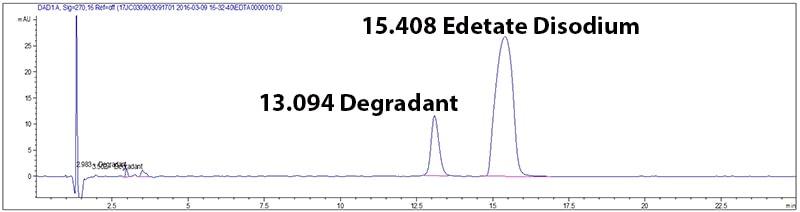
Figure 1: Example of a chromatogram
If the method is poorly designed, step one does not occur and the chromatogram displays only one peak. When the two different compounds are combined into one peak, the test provides an incorrect potency value. An incorrect potency result would lead to an inappropriate BUD assignment. To avoid this, all ingredients in the compounded preparation and drug degradants must be separated from the drug. Potency methods are designed to separate other ingredients in a drug product, but only stability indicating methods are designed to separate degradants.
Generation of degradants during method development
To develop a Stability-indicating method, ARL’s chemists create degradants that are likely to be formed as the compounded preparation ages. This is done through forced degradation studies. These studies involve subjecting the drug product and placebo to multiple stress conditions as recommended by these institutions:
- USP in USP<1225>: Validation of Compendial Procedures
SPECIFICITY
“If impurity or degradation product standards are unavailable, specificity may be demonstrated by comparing the test products to a second well –characterized procedure (e.g., a Pharmacopeial or other validated procedure). These comparisons should include samples stored under relevant stress conditions (e.g., light, heat, humidity, acid/base hydrolysis, and oxidation). - The FDA in FDA Guidance for Industry Analytical Procedures and Methods Validation
Stress Studies: “Degradation information obtained from stress studies (e.g., products of acid and base hydrolysis, thermal degradation, photolysis, oxidation) for the drug substance and for the active ingredient in the drug product should be provided to demonstrate the specificity of the assay and analytical procedures for impurities […] do not interfere with the quantitation of the active ingredient.” - ICH in ICH Validation of Analytical Procedures Text and Methodology Q2(R1)
1.2.2 “Specificity may be demonstrated […]. This should include samples stored under relevant stress conditions: light, heat, humidity, acid/base hydrolysis and oxidation.”
Per these organizations, ARL performs six forced degradation studies when developing stability indicating methods. Those are:
- Acid hydrolysis
- Base hydrolysis
- Thermal degradation
- Humidity
- Photolysis
- Oxidation
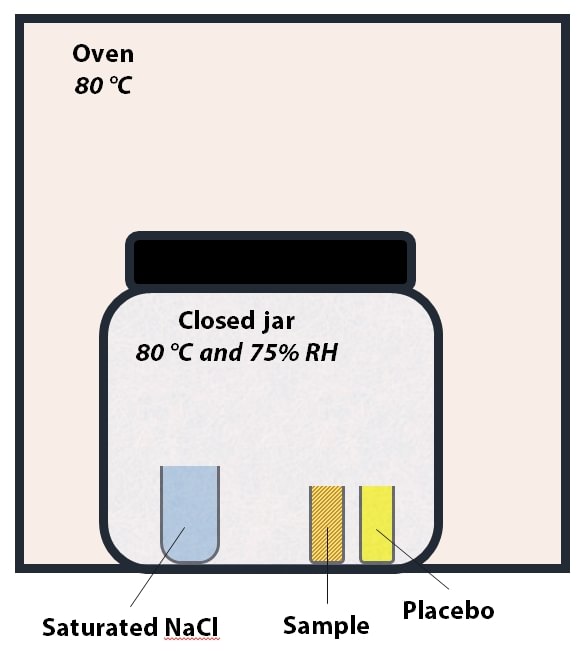
Figure 2: Set up for Heat and Humidity stress. The temperature inside the jar is 80 °C and relative humidity is ~75%
Once all stress conditions are performed, each stressed sample and controls are tested on the HPLC to see what degradation peaks are created. It is critical to examine each stress condition because different stress conditions can create different degradants. Notice in Figure 3, UV light stress generated different degradants than heat and humidity stress. The different degradants are circled in red.
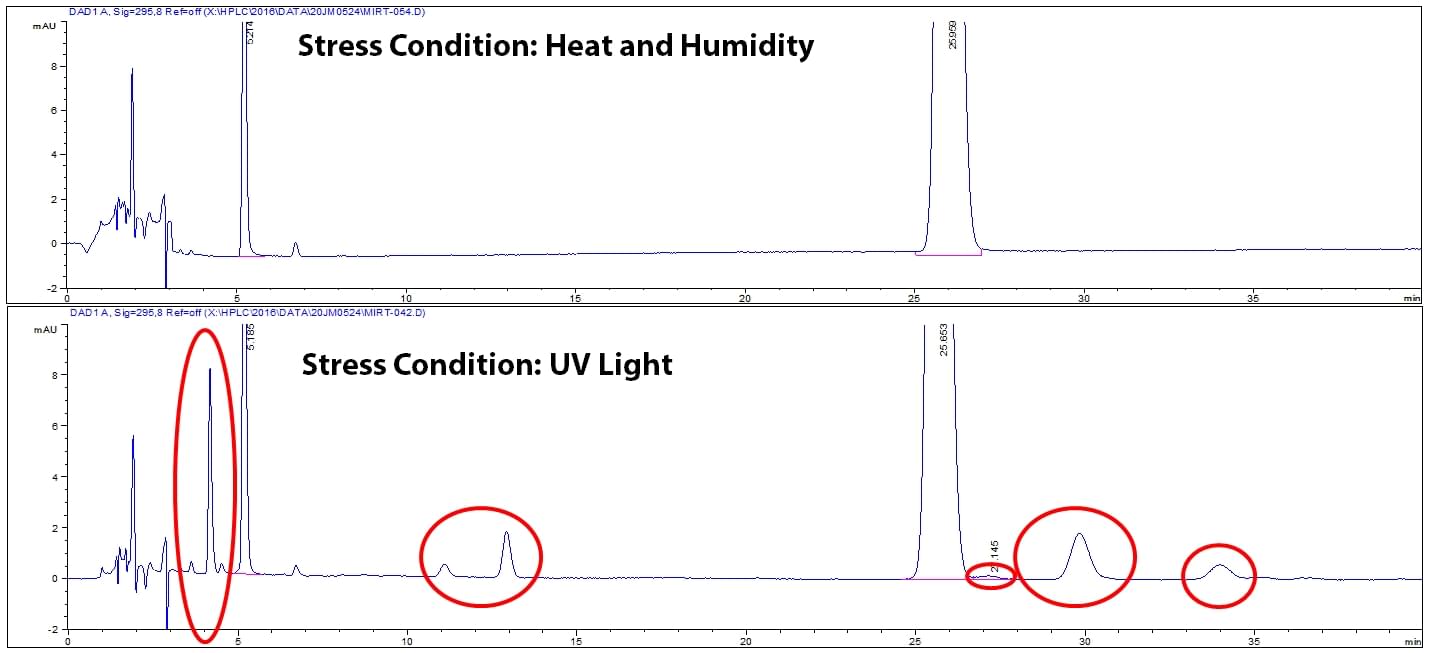
Figure 3: Example of degradation chromatograms for two stress conditions.
ARL chemists develop a test method that separates all degradants created by the stress studies from the drug. Once this separation is achieved, the method is considered stability indicating and can now be validated. Method validation ensures the test method is accurate, precise, has a linear response and is robust to small changes. After the stability indicating test method is validated, the test method is considered reliable, meaningful, and specific and the stability study can be conducted to determine the stability and beyond-use date of a product.
James Zellner, Technical Sales
Identification of a contaminant in a sterile preparation is vital to determining the origin of the microorganism and its avenue of entry. The organism’s identity, combined with environmental monitoring data, provides actionable information to improve a pharmacy’s processes and prevent future contamination.
Organisms can enter a drug product through:
- People
- Process
- Environment
The source of microorganisms can potentially be determined by considering their normal habitat. For example, a large group of organisms are considered normal skin flora. Microorganisms like Staphylococcus aureus, Micrococcus luteus, and Propionibacterium acnes are found on every technician that enters a cleanroom. If skin flora is found in a sterile preparation, it could indicate improper gowning and gloving or insufficient preparation of supplies and materials handled by personnel brought into the controlled area.
Another common organism group recovered from sterility samples and controlled areas are soil organisms. This includes members of the bacterial genus Bacillus and fungal organisms like Aspergillus and Cladosporium. If these microorganisms are found, it may indicate improper cleaning procedures, technicians tracking in contaminants on their shoes from outdoors, or failure to properly seal a controlled area, causing dust and debris from outside to be drawn in.
Water-borne organisms may also be found in compounded products. This list includes E. coli and Enterococcus. Recovery of these genera indicates a contaminated water source near the controlled area, or the water used to clean or compound is not suitable for the process.
Knowing the identity of the organism is the only way to determine how the contamination occurred, where the contaminant came from, and how to prevent it from entering a future drug product. Pharmacists can determine the likely origin of an identified organism by testing and consulting literature or a trained microbiologist. The specific identity of microorganisms recovered from a contamination also assists pharmacists in the development, regular use, and continued improvement of cleaning and sanitization plans. Compounders and testing labs must work together to generate reliable sterility testing results and examine test failures to ensure quality end products for patients.