
Labeling Requirements for Compounded Preparations
Labeling is an essential quality requirement for compounded preparations. Labeling procedures must comply with the laws and regulations of the applicable regulatory jurisdiction and must be included in the facility’s standard operating procedures. Each container must be clearly labeled with accurate and legible identifying information to prevent errors during storage, dispensing, and use. Additionally, the label must be verified to ensure it matches the prescription or medication order, the master formulation record, and the compounding record.
According to USP 795 and 797, the label on each container must, at a minimum, include:
- Assigned internal identification number (barcode, prescription, order, or lot number)
- Active ingredient(s), amounts, activities, and concentrations
- Beyond Use Date
- Compounding facility name and contact information if the preparation is to be sent outside of the facility or healthcare system in which it was compounded
- Dosage Form
- Indication that the preparation is compounded
- Route of administration
- Storage conditions if other than controlled room temperature
- Special handling instructions
- Total amount or volume if it is not obvious from the container
- Warning statements, if applicable
Sterile preparations must also include whether it is a single-dose or multi-dose container, if applicable.
According to the Food and Drug Administration, 503B facilities must ensure the label also includes:
- Date the drug was compounded
- List of active and inactive ingredients, identified by established name and the quantity or proportion of each ingredient
- National Drug Code, if available
- Statement “Not for resale”, and, if the drug is dispensed or distributed other than pursuant to a prescription for an individual identified patient, the statement “Office Use Only”
A final check must be conducted and documented to verify that the correct label has been affixed to the finished compounded drug product before dispensing. Federal and state regulations may require additional label information before dispensing a compounded preparation to a specific patient.
For more resources on labeling, please refer to:
- USP 7 Labeling
- USP 795 Pharmaceutical Compounding – Nonsterile Preparations
- USP 797 Pharmaceutical Compounding – Sterile Preparations
- Food and Drug Administration 21 U.S.C. § 353b(a)(10) (2022)
- Food and Drug Administration Current Good Manufacturing Practice – Guidance for Human Drug Compounding Outsourcing Facilities
- ASHP Guidelines on Compounding Sterile Preparations
Labeling Requirements for Compounding Animal Drugs
The labeling requirements for compounding animal drugs varies based on patient specific nonfood-producing animals, office stock for nonfood-producing animals, and antidotes for food-producing animals or sedatives, and anesthetics for free-ranging wildlife.
According to the Food and Drug Administration, compounding animal drug labeling must include:
- Beyond Use Date
- Drug name
- Name, address, and contact information for the compounding pharmacy or veterinarian and name of prescribing veterinarian
- Patient species
- Patient identification, name of patient, identifier for individual animal, or identification of a group of animals
- Strength
- Statement, “Report suspected adverse reactions to the [pharmacist or veterinarian who compounded the drug] and to FDA using online Form FDA 1932a”
- Statement, “This is a compounded drug. Not an FDA approved or indexed drug.”
- Statement, “Caution: Federal law restricts this drug to use by or on the order of a licensed veterinarian.”
For office stock for nonfood-producing animals, labeling must also include:
- Indication for which the drug will be used
- Name, address, and contact information for the veterinarian ordering the office stock
- Statement “Not for use in food-producing animals”
For antidotes for food-producing animals or sedatives and anesthetics for free-ranging wildlife, labeling must also include:
- Indication for which the drug will be used
- Name, address, and contact information for the veterinarian ordering the antidote, or the wildlife health professional ordering the sedative or anesthetic
- Prescribing veterinarian-determined withdrawal time
While lot numbers are not required on the labeling by the FDA for compounded animal drugs, it may be required by state law and is recommended in the case of a recall.
For more resources on compounding animal drug labeling, please refer to:
- Food and Drug Administration Compounding Animal Drugs from Bulk Drug Substances – Guidance for Industry #256

Sterility testing of compounded sterile preparations is crucial to prevent patient harm from microbial contamination. USP 797 requires sterility testing for:
- Category 2 CSPs which are assigned a beyond use date (BUD) that requires sterility testing.
- All category 3 CSPs.
The Food and Drug Administration (FDA) requires 503B outsourcing facilities to perform sterility testing for all drugs reported to be sterile and/or non-pyrogenic.
Sterility testing must be performed according to USP 71 or a validated alternative method per USP 1223 that is non-inferior to USP 71. Method suitability must be demonstrated for each drug product with correct sample quantities tested according to the chapter. Sterility failures must be investigated and include the identification of contaminating microorganisms with documentation of the investigation or corrective action.
Alternative Sterility Testing Methods
Rapid Sterility testing allows for shortened incubation times compared to the traditional sterility test method. By reducing sterility test wait times, rapid sterility methods offer many supply chain advantages, faster product release schedules, and quick contamination investigations.
ARL now offers two alternative sterility test methods to help minimize the impact of USP 797 shorter beyond-use dates.
ScanRDI Solid Phase Cytometry Method: 0-2 Day Turnaround
This non-growth-based sterility test detects microbial contamination based on cytometry, which detects and quantifies microorganisms in as little as four hours. This ultra-rapid microbial detection method shortens investigation time by 1-2 weeks, leading to faster quality control improvements and process normalization. Speed up your distribution with ScanRDI and release drug products more quickly, lowering costs and reducing waste while eliminating bottlenecks.
Celsis ATP Bioluminescence Method: 6 Day Turnaround
This growth-based sterility test detects microbial contamination based on the presence of microbial Adenosine Triphosphate (ATP) in a sample. Results are objective and based on instrument analysis. This system is flexible for filterable, nonfilterable, cloudy, and other complex products. The test is non-destructive, allowing for the identification of any microbes discovered to help kick-start your investigation.
USP 1223 Method Validation
ARL’s rapid sterility validation protocol includes all required parameters listed in USP 1223, including specificity, limit of detection, repeatability, and equivalency. For the growth-based method, the recovery of stressed/injured microorganisms is also assessed, which is considered imperative as injured microorganisms are more representative of potential contaminants that may be present in preparations tested.
Traditional Sterility Testing Method
USP 71 Sterility Test Method: 14-18 Day Turnaround
This sterility method is a growth-based test method that requires at least 14 days of incubation time for microorganism growth to occur. ARL uses either a closed membrane filtration method (the preferred method) or a direct inoculation method for unfilterable sample types. At the conclusion of the test, if there is no evidence of growth, the drug product complies with the USP 71 test for sterility.

Standard Operating Procedures for Compounding Pharmacies
Standard Operating Procedures (SOPs) are an essential component of quality systems. The United States Pharmacopeia (USP) states that SOPs must be developed for all aspects of the compounding operation. All personnel who conduct or oversee compounding must be trained in the facility’s SOPs and are responsible for ensuring the facility’s SOPs are followed. One or more persons must be designated to ensure that SOPs are fully implemented, and the designated person must ensure that follow-up occurs if problems, deviations, or errors are identified.
It’s important to review state board of pharmacy laws and regulations, USP chapters, and Food and Drug Administration (FDA) guidelines to determine SOPs for quality systems. The American Society of Health-System Pharmacist Compounding Resource Center provides SOP lists to help pharmacies comply with compounding standards.
USP 1029 Good Documentation Guidelines provide sections typically included in an SOP:
- Purpose and scope
- Instructions and procedure
- Responsibilities and roles
- Materials or equipment, as appropriate
- Definitions or references, as needed
- Review and approval
- Revision History
Additionally, ensure an SOP has a document number, effective date, and an approval signature.
When writing procedures, it’s important to consider the personnel’s perspective. Ensure procedures are clearly written in a step-by-step format. Avoid ambiguity and be careful with important terms. Remember, United States Pharmacopeia defines “should” as a recommendation and “must” as a requirement.
Testing Standard Operating Procedures for compounding SOPs, include but not limited to:
- Media Fill Testing
- Environmental Controls
- Total Airborne Particle Sampling
- Microbiological Air and Surface Monitoring
- Components
- Verifying the effectiveness of cleaning and disinfecting activities
- Verifying the effectiveness of sterilization and depyrogenation methods
- Release Inspections and Testing
- Quality Assurance and Control (out of specifications, complaints, and adverse event reporting)
- Hazardous Drug Testing/Sampling
The state boards of pharmacy and FDA will inspect to ensure that a pharmacy follows written procedures. Having an SOP review schedule and training personnel according to SOPs is essential. Changes to SOPs must be documented and communicated to all personnel involved in the process and procedure with acknowledgment documented.
For additional information on SOPs, access:
- USP 795 Pharmaceutical Compounding Nonsterile Preparations
- USP 797 Pharmaceutical Compounding Sterile Preparations
- USP 800 Hazardous Drugs Handling in Healthcare Settings
- USP 1029 Good Documentation Guidelines
- USP 1163 Quality Assurance in Pharmaceutical Compounding
- USP Compounding Compendium – Access Table of Contents

Media fill testing is a crucial part of personnel qualification and environmental monitoring. During a media fill, technicians use microbiological growth media instead of a drug solution to assess whether the aseptic procedures performed are adequate to prevent contamination during sterile compounding.
USP 797 requires media fill testing before beginning to compound Category 1, 2, or 3 CSPs; and:
- Once every six months for Category 1 and 2
- Once every three months for Category 3
USP 797 also requires annual media fill testing for designated personnel. If a designated person also compounds, the individual must complete the aseptic manipulation competency at the same intervals required for compounding personnel.
Media Fill Design
Media fill simulates the most challenging compounding procedures and captures process areas that could potentially affect the sterility of CSPs, including:
- Factors associated with the length of the process that can pose contamination risk (e.g., operator fatigue, quality of equipment)
- Number of aseptic additions or transfers
- Number, type, and complexity of manipulations
- Number of personnel in the buffer room or SCA
Simulations should reflect the compounding activities conducted inside the Primary Engineering Control (PEC). If all starting components are sterile, simulate a sterile-to-sterile compounding activity. If some starting components are non-sterile, simulate a non-sterile to sterile compounding activity. Follow the media fill testing procedures outlined in USP 797, which can be accessed through the USP Compounding Compendium.
Once the compounding simulation is complete and the final containers are filled with the test media, perform a gloved fingertip and thumb sample on each hand and surface sample of the direct compounding area inside the PEC. Take the samples prior to disinfecting gloves and PEC. Handle and store samples to avoid contamination and incubate inverted to prevent condensate from dropping onto the agar during incubation and affecting the accuracy of the colony-forming unit (CFU) reading.
Incubate the final containers at 20°-25° C and 30°-35° C for a minimum of 7 days at each temperature to detect a broad spectrum of microorganisms. The order of the incubation temperatures must be described in the facility’s standard operating procedures (SOPs). Final containers must be incubated in an incubator. A media test failure is indicated by visible turbidity or other visual growth in the media in one or more container closure unit(s) on or before 14 days.
Incubate plates at 30°-35° C for no less than 48 hours and examine for growth. An actionable level for gloved fingertip and thumb sampling after media fill testing is >3 CFU, total from both hands.
Growth Media
The most common growth media is soybean casein digest medium (SCDM), which is also known as Trypticase Soy Broth (TSB). Growth media may be obtained from a commercial supplier or prepared in-house:
- If using a commercial supplier, a certificate of analysis (COA) must be obtained and state that the lot of growth media supports the growth of microorganisms. Suppliers are qualified by testing three batches of media using quality control tests: growth promotion, pH, sterility, and visible inspection.
- If preparing growth media in house, growth promotion of the media must be demonstrated for each batch and documented as described in Sterility Tests USP 71.
Media Fill Controls
A media fill is an experiment that should include controls. These controls should be independent of the growth promotion testing.
A positive control is a sealed product container of media inoculated with a small number of microorganisms. If growth occurs, the positive control demonstrates that the media can promote the growth of the organism(s). The positive control should be inoculated in an area separate from the compounding area.
A negative control is prepared by transferring media into a separate sterile container and incubating the control with the media fill test containers. The negative control should not show microorganism growth proving the media was sterile at the start.
ARL Services
ARL provides testing services to assist compounders with personnel qualification and environmental monitoring.
- Growth Promotion – this test demonstrates that the media used in environmental monitoring programs, media fills, or personnel qualification are capable of supporting microorganism growth. ARL tests according to client instructions and provides a certificate of analysis for specific microorganisms.
- Incubation / Observation – this test incubates and observes the media and plate samples according to client instructions and provides a certificate of analysis with CFU count.
- Microbial Identification – this test identifies microorganisms isolated during incubation and observation and tests the genus/species level using DNA sequencing.
Contact ARL for more information on testing at 800-393-1595 or info@arlok.com.

Kathy Heatherly, MSFS, ARL Associate Chemistry Supervisor
A bulk substance, or active pharmaceutical ingredient (API), is often the starting point of a compounded preparation. Compounding pharmacies and outsourcing facilities should verify the quality of the API before use, ensuring it meets industry standards, and the component is suitable for use in a compounded preparation. At a minimum, identity testing is recommended for each lot, with additional rigorous testing as necessary.
Guidelines for Testing Active Pharmaceutical Ingredients
The United States Pharmacopeia (USP), Food and Drug Administration (FDA), and International Council for Harmonization (ICH) provide guidelines for testing Active Pharmaceutical Ingredients (APIs).
- USP General Notices, section 4.10.10, states a single monograph may include multiple tests and acceptance criteria for the same attribute. Unless otherwise indicated in the monograph, all tests are required and may vary based on different manufacturers’ product attributes, such as polymorphic forms, impurities, hydrates, and dissolution.
- cGMP Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act Guidance for Industry, states components that are not approved finished drug products must be tested to verify identity and evaluated for conformity with appropriate specifications.
- ICH Good Manufacturing Practice for Active Pharmaceutical Ingredients (API) Q7 provides guidelines to ensure APIs meet the quality and purity claims of the manufacturer. Sections 7.30 – 7.32 state that at least one identity test must be conducted for each batch, except for specified materials. A supplier’s Certificate of Analysis (CoA) may substitute for additional tests if the manufacturer has a system to evaluate and confirm that suppliers consistently meet specifications. At a minimum, a full analysis should be conducted at appropriate intervals and compared with the CoA. The reliability of these certificates should also be regularly checked.
Compendial Test Methods
Testing for potency, sterility, and endotoxins on the finished product is not enough to determine the API’s suitability for compounding. Similarly, a test that verifies only the API’s identity may not ensure its fit for use in the final product. Additional compendial test methods that should be evaluated include:
- Organic impurities
- Inorganic impurities
- Enantiomeric purity
- Residual Solvents
- Volatile Matter
- Microbial Enumeration
- Assay
Many of the USP monographs provide specifications for calculating the assay value that should be used when compounding a finished preparation. If the substance is a hydrate, its anhydrous equivalent weight may need to be calculated. On the other hand, if there is adsorbed moisture present, the weight of anhydrous drug substance may need to be calculated. In addition to testing when the material is sourced, if there have been multiple entries into the container over the life of the material, it is also recommended to test the moisture content again; as additional moisture can affect the potency of the finished product.
Verifying Drug Components
Verifying that the correct material is sourced and that the specifications are met is essential. This helps ensure product integrity and quality in the finished product.
503A Pharmacies can verify drug components by:
- Reviewing the supplier’s certificates of analysis (COA)s;
- Establishing the reliability of the supplier’s analyses through vendor qualification; and,
- Testing drug components before use in compounding.
503B outsourcing facilities can verify drug components by:
- Testing each shipment of the ingredient; or
- In lieu of testing each shipment, a COA can be accepted from the supplier and evaluated to determine whether a lot can be used, provided:
- Pharmacists confirm the supplier’s test results, no less frequently than annually for active ingredients and every two years for other components, by performing full compendial testing using the applicable USP or NF monographs and/or supplier’s in-house methods.
- Pharmacists conduct at least one identity test to confirm the component purchased from the supplier.
For more information on bulk drug testing, contact ARL at 800-393-1595 or info@arlok.com.
Kayla Lipcaman, ARL Bio Pharma Associate Microbiology Supervisor
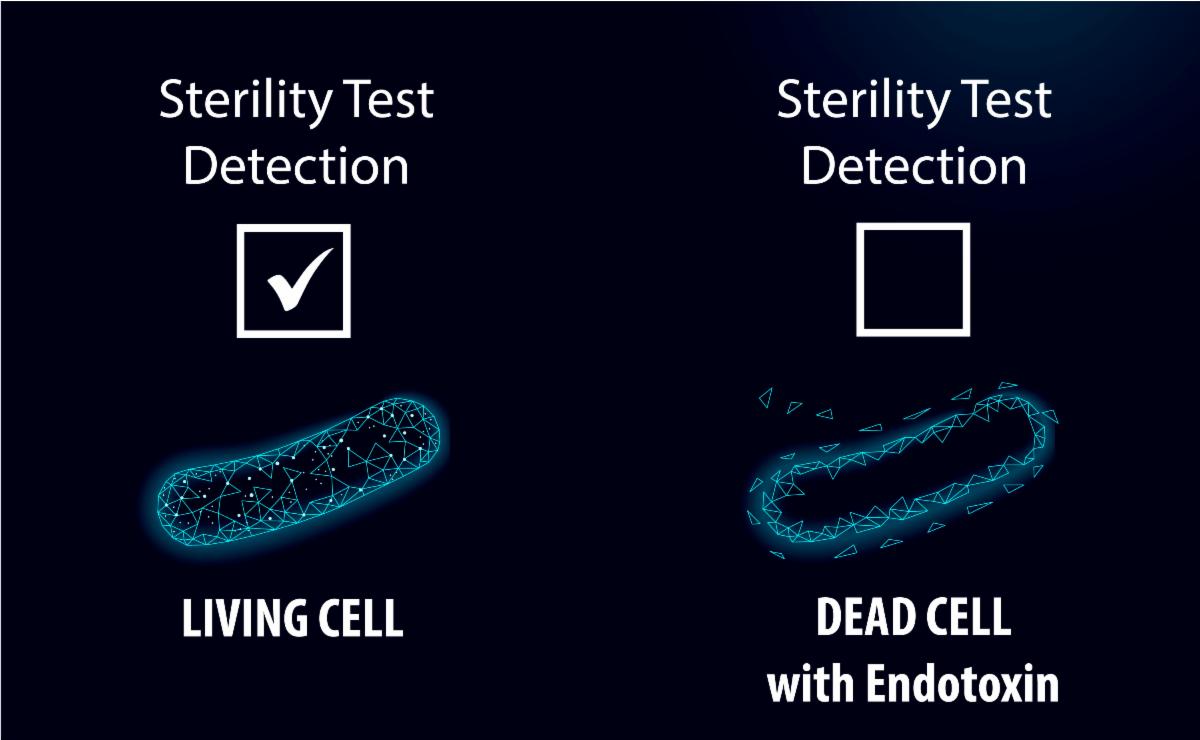
Two critical tests for sterile compounded preparations are Sterility and Endotoxin testing.
USP 797 requires Sterility testing for:
- Category 2 CSPs assigned a beyond-use date that requires sterility testing
- Category 3 CSPs
USP 797 requires a Bacterial Endotoxin Test (BET) for:
- Category 2 and Category 3 CSPs compounded from one or more nonsterile component(s)
- Multiple-dose CSPs
Sterility testing is a fundamental aspect of quality assurance for compounded pharmaceuticals, ensuring that samples tested are free from viable microorganisms. This is crucial for patient safety, as contaminated products can lead to serious health complications. Sterility testing is a control check meant to assess whether the compounding process has been performed in a manner that limits contamination, indicating the product is safe for use. It is also a critical component of many regulatory standards set forth by organizations like the FDA, USP, and State Boards of Pharmacy.
Importantly, a passing sterility test alone does not guarantee that a compounded product is completely safe. While sterility testing is meant to detect viable microorganisms, it is not designed to identify harmful bacterial byproducts, such as endotoxins. Endotoxins are toxic substances released from the outer membranes of certain bacteria, particularly Gram-negative bacteria, when they die or break apart. Even if a product is sterile and free from living bacteria, endotoxins may still be present and can result in dangerous immune responses in patients, including fever, inflammation, and septic shock. This is especially concerning when products are introduced into the bloodstream. A ‘Sterile’ test result does not ensure that a compounded product is free from these toxins. Given this, a robust testing regimen often includes both Sterility and Endotoxin testing, to provide assurance that a product meets patient safety expectations. By performing both Sterility and Endotoxin testing, a pharmacy is ensuring that their compounded medications undergo comprehensive safety assessments. This testing strategy provides ARL’s clients with confidence that the products are safe from both microbial contamination and the toxic effects of endotoxins, thus maintaining the highest standards of product quality and patient care.
For more information on sterility and endotoxin testing, contact ARL at 800-393-1595 or info@arlok.com.

What is stability?
The extent to which a product or preparation retains physical and chemical properties and characteristics within specified limits throughout its expiration or beyond use date (BUD).
When is stability data required?
Compounders must consider the chemical and physical stability of the compounded preparation and its components when establishing a BUD. Stability data utilizing a stability-indicating analytical method is required to:
- Determine if a shorter BUD must be assigned when the stability is less than the limits specified in USP 795 and 797
- Extend BUD for nonsterile preparations up to a maximum of 180 days
- Support BUD for Category 3 sterile preparations
What tests are included in a stability study?
Tests are based on dosage form, formulation, and product properties and demonstrate chemical, physical, and microbiological stability. Tests may include:
- Sterility (for sterile preparations)
- Endotoxin (for most sterile preparations)
- pH
- Appearance
- Particulate matter
- Potency/ Assay
- Antimicrobial effectiveness testing for aqueous preparations
- Preservative quantification
- Microbiological tests for water-containing formulations
- Microbial Enumeration Tests (for nonsterile preparations)
- Tests for Specified Microorganisms (for nonsterile preparations)
- Container closure integrity
How many time points are included in a stability study?
The timing for conducting tests is determined based on the desired beyond-use date. ARL recommends conducting tests at the beginning, end, and at least three time points. Not all tests need to be conducted at every time point, but doing so allows for a more comprehensive analysis of the preparation’s stability and provides intermediate time points to use if a test result is out of specification. The stability design may vary depending on the test type and the frequency required by USP guidelines.
Stability Study Design Example:
| Test | Method Type | Time Points (Days) | # of Time Points |
|---|---|---|---|
| Appearance | Visual | 0, 30, 60, 90, 180 | 5 |
| pH | USP 791 | 0, 30, 60, 90, 180 | 5 |
| Particulate Matter | USP 788 | 0, 30, 60, 90, 180 | 5 |
| Potency Assay | HPLC | 0, 30, 60, 90, 180 | 5 |
| Sterility | USP 71 | 0, 90, 180 | 3 |
| Endotoxin | USP 85 | 0, 180 | 2 |
How long does a stability study take?
The stability study turnaround time depends on the stability-indicating analytical method. For a method to be considered stability-indicating, the method must be developed and validated per USP 1225. The validation includes accuracy, linearity, precision (repeatability), range, specificity, and system suitability. Specificity demonstrates non-interference from impurities and matrix components and involves stress studies/ forced degradation to demonstrate the method is capable of separating degradation products from the active pharmaceutical ingredient (API).
Stability Indicating Analytical Method Options
Method Development and Validation: ARL develops and validates a stability-indicating analytical method for the quantitation of the API(s). After the method is validated, ARL evaluates the physical, chemical, and microbiological properties of the packaged product over a target expiration period.
Method development and validation must be performed on the exact product being tested to eliminate interference from degradants and impurities.
Development Time: 8 – 12 weeks (time varies based on formulation complexity)
Method Verification: ARL verifies a previously validated stability-indicating analytical method is appropriate for the quantitation of the API(s). The laboratory demonstrates specificity for matrix components, system suitability, and accuracy of the previously validated method. After the method is verified for the formulation, ARL evaluates the physical, chemical, and microbiological properties of the packaged product over a target expiration period.
Method verifications can be used on the same or similar products tested with fewer APIs and/or excipients.
Verification Time: 4 – 6 weeks
Method Applicability Assessment: ARL compares the drug product being tested to a previously validated stability-indicating analytical method and performs system suitability analysis. A successful system suitability analysis indicates no difference between the product being tested and the previously validated formulation.
Applicability Time: 4 weeks (time varies based on tests performed and GMP vs. non-GMP)
When are stability protocols required?
Protocols for method work and stability are required for GMP stability-indicating analytical methods. The protocol directing the method work will specify the methods and acceptance criteria used to validate the method. The stability study protocol will specify the tests, methods, timepoints, and acceptance criteria applied during the stability study.
What is ARL’s wait time to start method development, verification, or assessment?
ARL’s wait time fluctuates with the seasons and demand. To avoid delays, we highly recommend submitting a quote request ahead of time and preparing for a minimum 12-week wait.
Currently, there is no wait time for ARL to start work on your method.
Contact ARL for more information on stability studies info@arlok.com or 800-393-1595.

Bracketing and its application to quality testing
Bracketing is a testing design approach recognized by USP, FDA, and ICH for assessing the quality of pharmaceutical products. It involves testing certain quality attributes at the lowest and highest extremes and assumes that intermediate levels are represented by the extreme test values. This design strategy reduces the samples needed for testing while ensuring product integrity.
Bracketing can be applied to compounded drug products with multiple strengths of identical or closely related formulations. Bracketing can also be applied to formulations of the same container closure system, where either container size or fill varies while the other remains constant. If both container size and fill vary, it’s critical not to assume that the largest and smallest containers represent the extremes of all packaging configurations. Compounders must carefully select the extremes by comparing container closure system characteristics that may affect product stability.
Examples where bracketing may be applied to drug products with one variable change:
| Drug Product | Formula Strength | Container | Container Size | Fill Volume |
|---|---|---|---|---|
| Injection / IV Solution | Different | Same | Same | Same |
| Injection / IV Solution | Same | Same | Different | Same |
| Injection / IV Solution | Same | Same | Same | Different |
| Ophthalmic Solution | Different | Same | Same | Same |
Bracketing should not be applied in cases where:
- Different excipients are used among strengths
- Strengths, container sizes, and/or fills selected for testing are not the extremes
Bracketing in a Stability Study Design
Bracketing is a risk-based approach and requires scientific justification before implementation. Considerations include, but are not limited to:
- Product Characteristics
- Regulatory Requirements
- Test Parameters
- Data Analysis
ICH Harmonized Guideline for Bracketing and Matrixing Designs for Stability Testing of New Drug Substances and Products, Table 1 provides an example of a product available in three strengths and container sizes. The 15 mL and 500 mL container sizes represent the extremes, and batches for each selected combination should be tested at each time point, similar to a full design.
| Strength | 50 mg | 75 mg | 100 mg | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Batch | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| Container size | 15 ml | T | T | T | T | T | T | |||
| 100 ml | ||||||||||
| 500 ml | T | T | T | T | T | T | ||||
If a product has different storage conditions, each storage condition should be treated separately under its design.
Compounders must thoroughly evaluate the bracketing stability data before applying a beyond-use date. This assessment should consider the stability of the extremes. If they differ, it is important to consider the intermediates no more stable than the least stable extreme.
To request a bracketing stability study design, request a quote.
Additional Bracketing Applications
Bracketing may also be applied to tests and method validations with scientific justification.
Antimicrobial Effectiveness Testing:
Both 795 and 797 state antimicrobial effectiveness testing must be conducted and passed once for each formulation in accordance with USP 51 for the particular container closure system in which it will be packaged. A bracketing design may be used to test a low concentration and a high concentration of the active ingredient in the formulation to establish preservative effectiveness across various strengths of the same formulation. The concentration of all other ingredients (including preservatives) must be the same throughout the bracketing study.
ARL requires method suitability for each formulation tested.
Endotoxin Method Validation:
Method validation is recommended to ensure there is no inhibition or enhancement of endotoxins. When multiple concentrations of the sample product family are received, a bracketing design may be performed during validation. ARL will test the highest and lowest extreme concentrations. If an appropriate method for both formulations can be identified where the test method is the same, intermediate products can be referenced using the same test method.
For more information on bracketing, contact ARL at 800-393-1595 or info@arlok.com.

It is important to send the appropriate sample size for testing in order to obtain accurate and reliable results. The required sample quantity may vary depending on the formulation and specific test performed. Additionally, when sending in samples for microbiology testing, it is crucial to take into account the batch size, as sampling quantities are determined based on statistical probabilities of identifying contamination across different articles and batch sizes.

Sampling is formulation dependent.
Minimum sample size for each active ingredient tested:
- Raw powder: 50 mg
- Triturates/Granulations/Blend Powders: 1 g
- Solid Dose Forms (Tablets, Capsules, Pellets,
- Troches, Suppositories): 5 units
- Suspensions: ≥10 mL, recommend ½ container size
- Solutions: 2 mL
- Semi-Solid Dose Forms (Creams/Lotions/Gels): 3 g
Read more about the Importance of Sample Amounts

Sampling is based on the method and is dependent on the formulation and dosage form.
USP Requirements:
- Therapeutic proteins: 1.5 mL
- Parenterals, topical and intraocular ophthalmics, or injections:
- If fill volume is <25 mL – 10 containers
- If fill volume is ≥ 25 mL – 1 container
Please provide 30 mL if using 1 container. The laboratory takes 25 mL for testing. Providing 30 mL ensures an ample sample amount for the test method.

Sampling is dependent on batch size and dosage form.
- USP 71 – Reference Table 3 in the chapter
- USP 797 – For batches <40 articles, the sampling size must be the number of units equal to 10% of the number of CSPs prepared, rounded up to the next whole number.
Sampling for method suitability:
- Sterility – 3x the certification amount
- Rapid sterility – 4x the certification amount

Sampling is based on the method.
- 1 mL or 100 mg
- Medical Devices: 10 articles
Sampling for method validation: 5 mL or 250 mg

Sampling is based on the method with enough sample to add large amounts of microorganisms into the preparation and simulate a contamination event during or after compounding.
- 70 mL or 70 g
Method suitability and testing can be conducted from the same 70 mL or 70 g supplied.
Sampling is based on the method, with enough sample to be plated on two types of growth media.
- 12 mL or 12 g
Method suitability and testing can be conducted from the same 12 mL or 12 g supplied.

SampSampling is based on the microorganism tested
- 12 mL or 12 g
Method suitability and testing can be conducted from the same 12 mL or 12 g supplied.
- Candida albicans – 2 mL or 2 g
- Clostridia sporogenes – 4 mL or 4 g
- Escherichia coli – 2 mL or 2 g
- Pseudomonas aeruginosa – 2 mL or 2 g
- Salmonella enterica – 12 mL or 12 g
- Staphylococcus aureus – 2 mL or 2 g
- Bile-Tolerant, Gram-Negative – 2 mL or 2 g
The sampling size for method suitability is the same as the sampling size for the test.
For additional tests and sample size requirements, log in to ARL’s Client Portal and access our price book.

The United States Pharmacopeia requires each facility to have a designated person for chapters 795, 797, 800, and 825. This role is vital to maintaining a facility’s adherence to quality standards.
The designated person, or persons, may be designated for more than a single site and does not have to be a pharmacist or in an administrative position. This allows for a diverse range of personnel to take on this responsibility.
Designated Person Responsibilities include, but not limited to:
- Quality Assurance and Quality Control Programs
- Personnel Training and Evaluations
- Personal Hygiene and Garbing
- Facilities and Engineering Controls
- Environmental Quality and Control
- Microbiological Air and Surface Monitoring
- Equipment, Supplies, and Components
- Compounding
- Sterilization Methods
- Establishing Beyond Use Dates
- Complaints and Adverse Reporting
- Develop, Monitor, and Review Standard Operating Procedures
- Corrective Actions
- Documentation
- Drug Product Handling and Storage
- Drug Product Quality
- Ensure Compliance with the Chapter
American Society of Health-System Pharmacists provides a resource document charting designated person(s) responsibility assignment and chapter applicability. Click here to access.
ARL provides resource documents for:
- Personnel Qualification and Environmental Monitoring
- Drug Product Quality Tests for Sterile and Non-Sterile
- USP 800 Surface Wipe Sampling
- Establishing Beyond Use Dates
Each facility should subscribe to the United States Pharmacopeia Compounding Compendium and refer to its state board of pharmacy guidelines and accreditation entities for additional designated personnel requirements.
To request a quote for testing services to support your facility’s compliance with quality standards, contact info@arlok.com or 800-393-1595.
Resources: