Maintaining control of environmental conditions is crucial in a compounding pharmacy. All personnel who compound or supervise compounding personnel must undergo initial training and demonstrate competency in compounding sterile preparations and achieving and maintaining appropriate environmental conditions.
USP 797 classifies compounding sterile preparations (CSPs) into three categories based on the level of environmental control during compounding, the likelihood of microbial growth during storage, and the duration within which they must be used.
Category 1: Compounded under the least controlled environmental conditions. These preparations may be prepared in a Primary Engineering Control (PEC) located in an unclassified segregated compounding area. They are assigned a BUD (beyond-use date) of ≤ 12 hours at controlled room temperature or ≤ 24 hours when refrigerated.
Category 2: Require more environmental controls and testing than Category 1. These preparations must be prepared in a cleanroom suite. They may be assigned a BUD of > 12 hours at controlled room temperature or > 24 hours if refrigerated.
Category 3: Undergo sterility testing, supplemented by endotoxin testing when applicable, and have more requirements than Category 2 CSPs for personnel qualification, use of sterile garb, use of sporicidal disinfectants, frequency of environmental monitoring, and stability determination. They have additional requirements that must be met at all times. These preparations may be assigned a BUD longer than established for Category 2 CSPs, up to 180 days.
Before beginning to compound Category 1, 2, or 3 CSPs, personnel are required to successfully complete 3 separate evaluations in succession for hand hygiene, garbing, gloved fingertip and thumb sampling, media fill, and surface sampling.
USP 797 – Personnel Qualification and Environmental Monitoring Requirements (after successful completion of initial competency)
| Test | Category 1 and 2 | Category 3 |
|---|---|---|
| Visual observation of hand hygiene and garbing | Once every 6 months | Once every 3 months for personnel who compound Category 3 CSPs |
| Gloved fingertip and thumb sampling | Once every 6 months | Once every 3 months for personnel who compound Category 3 CSPs as part of garbing competency and aseptic competency |
| Media Fill Testing | Once every 6 months | Once every 3 months for personnel who compound Category 3 CSPs |
| Post media fill surface sampling | Surface sample of direct compounding area after each media-fill competency | Surface sample of direct compounding area after each media-fill competency |
| Surface sampling | Monthly | Weekly |
| Viable air sampling | Once every 6 months | Monthly |
Visual Observation of Hand Hygiene and Garbing: This is a competency evaluation of hand hygiene, garbing procedures, and gloved fingertip and thumb sampling of both hands.
Gloved fingertip and thumb sampling: This test provides data on any microorganisms present on the technician’s gloves during compounding. Send plates to ARL for incubation, counting colony forming units (CFUs), and a Certificate of Analysis.
Media Fill Testing: This test assesses the technician’s ability to compound aseptically. Send media fill containers to ARL for incubation and observation, and a Certificate of Analysis.
Post-media fill surface sampling test: This test assesses the technician’s ability to compound aseptically. At the completion of the media fill qualification process, send surface sample plates to ARL for incubation, counting colony-forming units (CFUs), and a Certificate of Analysis.
Surface sampling: This test monitors surfaces for viable particles. Send plates to ARL for incubation, counting colony-forming units (CFUs), and a Certificate of Analysis.
*For Category 3 CSPs, surface sampling must be completed before assigning a BUD longer than the established limits. Additionally, surface sampling must be conducted within the PEC used to prepare Category 3 CSPs, at the end of each batch before cleaning and disinfection occurs, unless a self-enclosed robotic device is used. When a self-enclosed robotic device is used as the PEC to prepare Category 3 CSPs, surface sampling must be conducted at least once daily at the end of compounding operations, before cleaning and disinfecting occurs.
Viable air sampling: This test monitors air quality for airborne particles. Send plates to ARL for incubation, counting colony-forming units (CFUs), and a Certificate of Analysis.
For more information on personnel qualification and environmental monitoring, contact ARL at info@arlok.com or 800-393-1595.
USP 795 – Personnel Qualifications
All personnel who compound or have direct oversight of compounding nonsterile preparations (CNSPs) must complete training and demonstrate knowledge of principles and competency of skills for performing nonsterile manipulations as applicable to their assigned tasks.
Knowledge and competency must be demonstrated initially and at least every 12 months in at least the following core competencies:
- Hand Hygiene
- Garbing
- Cleaning and Sanitizing
- Handling and transporting components and CNSPs
- Measuring and mixing
- Proper use of equipment and devices selected to compound CNSPs
- Documentation of the compounding process
In addition, a pharmacy’s quality assurance system must ensure that the compounding process consistently meets quality standards, including environmental quality and maintenance.
Resources:
- USP 797 – Pharmaceutical Compounding—Sterile Preparations
- USP 795 – Pharmaceutical Compounding—Nonsterile Preparations
- USP 1116 – Microbiological Control And Monitoring Of Aseptic Processing Environments
- USP 1163 – Quality Assurance In Pharmaceutical Compounding
- FDA Guidance For Industry Sterile Drug Products Produced By Aseptic Processing – Good Manufacturing Practice
Tue, 04/23/2024 – 12:00
Available Drugs:
- 5-Fluorouracil
- Anastrozole
- Carboplatin
- Chloramphenicol
- Cisplatin
- Cyclophosphamide
- Estriol
- Estradiol
- Estrone
- Finasteride
- Fluconazole
- Medroxyprogesterone Acetate
- Methimazole
- Methotrexate
- Mitomycin
- Misoprostol
- Progesterone
- Testosterone
- Testosterone Cypionate
- Testosterone Enanthate
- Testosterone Propionate
- Tretinoin
- Voriconazole
If you would like to test for a drug not listed, please email info@arlok.com and request that a new drug be added to our testing library.
Nicole Vu, PhD, Scientific Director, ARL Bio Pharma
Compounding with synthetic peptides
Therapeutic peptides are biosimilar molecules, composed of a-amino acid (AA) polymer with a defined sequence of not more than 40 AA covalently linked by amide bonds. These biosimilar molecules are crucial to various biological processes including hormone regulation, immune function, wound healing, and metabolisms. Many therapeutic peptides are designed to have structural characteristics essential for their pharmacological effect and stability in-vivo, but they are very sensitive to storage and handling conditions. Thus, these peptide molecules deserve special attention to maintain their quality attributes through their period of use for consistent results in compounded preparations.
Synthetic peptide as active pharmaceutical ingredient (API)
Unlike compendial articles, manufacturers of synthetic peptides use in-house developed processes to produce peptide drug substances, and the manufacturer’s certificate of analysis (COA) is the official COA for the material. Before using these substances in compounded preparations, compounding pharmacies and outsourcing facilities should verify their quality. This includes confirming the correct identity and other critical attributes of the material, which should be traceable to a trusted reference standard. When sending the API to a contract laboratory for re-qualification testing, it is crucial to follow the manufacturer’s instructions for shipping and storage conditions. Storing the peptide in a tightly closed container at ambient temperature will maintain its stability for days to weeks. For longer storage, it is recommended to keep the peptides in a dark, cool place.
In general, bulk powder is structurally porous due to the small particle size of crystals that make up the bulk. Thereby bulk powder has a large surface area and high potential for moisture absorption. In addition, peptide molecules contain many amino acid residues with polar hydrophilic groups that contribute to the hygroscopicity of the bulk material. Frequent exposure of peptide material to the ambient atmosphere will eventually lead to changes in its physicochemical properties, such as agglomeration, solubility, microbial spoilage, and reduced stability of the peptide molecules. Therefore, frozen or refrigerated materials should be at room temperature prior to compounding to avoid moisture condensation and absorption that could shorten the shelf life of these products.
Challenges in compounding with synthetic peptides
While the intramolecular structure and intrinsic properties of AA residues determine the chemical and physical stability of peptide molecules in compounded preparations, the compounding process itself can introduce conditions conducive to peptide instability. These conditions include mixing, trituration, high application temperature, out-of-range pH, and high concentration of peptide in the formula. As a result, one of the most common problems in compounding with biosimilar peptides is aggregation, which leads to the formation of dimers, trimers (oligomers), and sub-visible particles
In the absence of manufacturing controls, formulation excipients, solvents, and container/closure used in compounding can impact the stability, safety, and efficacy of peptide preparations. For example, peroxide in polysorbate 80, Shift-base reaction with dextrose, and metal leaches from glass or syringe containers and from rubber stoppers, etc. To ensure a quality drug, compounders should carefully select solvents, vehicles, and container/closure when compounding peptides, as they can impact the solubility, stability, and effectiveness of the medication.
Contact ARL to request a quote for testing peptides.
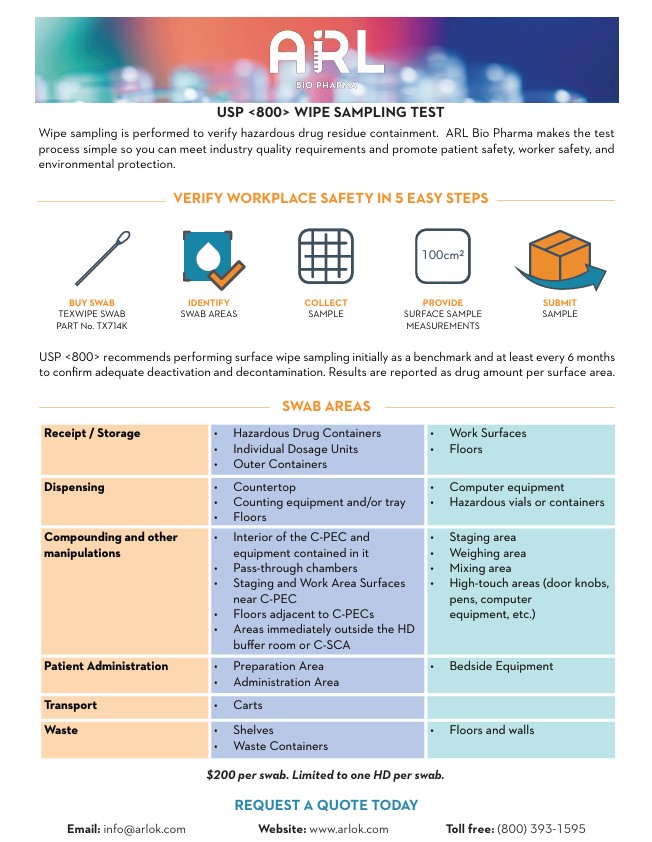
USP 800 is a quality standard that promotes patient safety, worker safety, and environmental protection when handling hazardous drugs (HDs). It applies to all healthcare personnel who handle HDs, and all entities that store, prepare, transport, and administer HDs.
As of November 1, 2023, USP 800 is required and compendially applicable to USP 795 and 797 when a practitioner is compounding HDs.
An HD is a drug identified as hazardous or potentially hazardous by the National Institute of Occupational Safety and Health (NIOSH).
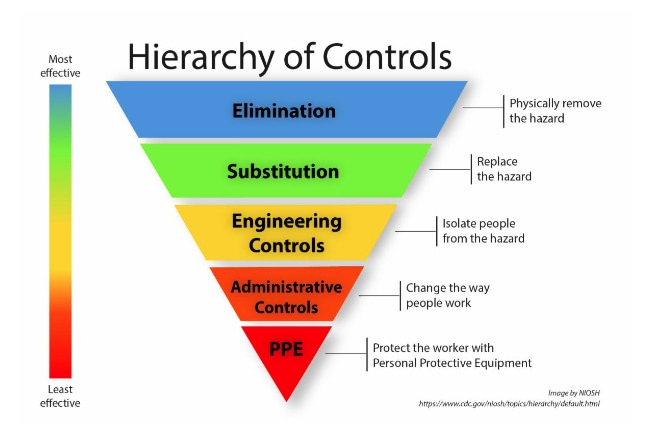
Exposure to HDs can occur through dermal, inhalation, ingestion, or injection. NIOSH published a hierarchy of controls with five actionable levels to reduce or remove hazards. In compounding, the first line of defense against HD exposure is in engineering controls within containment areas and hoods. The second line of defense is administrative controls, followed by personal protective equipment (PPE).
Pharmacies can determine the effectiveness of their HD engineering controls by performing surface wipe sampling to detect HD residue. USP 800 recommends performing sampling routinely, initially as a benchmark and at least every 6 months. Pharmacies may also perform these tests more frequently to ensure the effectiveness of containment strategies and work practices.
There is no pass rate for surface wipe sampling; however, an acceptable surface limit can be established from the occupational exposure limit in a pharmaceutical manufacturer’s safety data sheet.
The suggested sampling areas include the interior of the Containment Primary Engineering Control (C-PEC) and equipment in it, pass-through chambers, surfaces in staging or work areas near the C-PEC, areas adjacent to C-PECs, areas immediately outside the HD buffer room or Containment Segregated Compounding Area (C-SCA), and patient administration areas.
Pharmacies need to be prepared to evaluate and investigate contamination. USP states that if any measurable contamination is found, designated individuals must identify, document, and contain the cause of contamination. This can be achieved by reevaluating work practices, re-training personnel, performing thorough deactivation, decontamination, and cleaning, and improving engineering controls. It is important to repeat the wipe-sampling process to ensure that the deactivation, decontamination, and cleaning steps are effective.
To send a surface wipe sample to ARL for testing, follow these steps:
- Purchase a swab (TEXWIPE SWAB PART No. TX714K).
- Swab the area to be tested.
- Submit the swab for analysis. Please do not submerge the swab in any liquid.
The cost for testing is $200 per swab, and only one hazardous drug can be tested per swab.
Once the testing is complete, a certificate of analysis will be issued with a quantitative result based on the amount of the drug detected on the swab.
For more information on surface wipe sampling, contact ARL at info@arlok.com or 800-393-1595.
Resources:
- USP 800 Hazardous Drugs – Handling in Healthcare Settings
- National Institute for Occupational Safety and Health
- National Institute for Occupational Safety and Health Hierarchy of Controls
- ASHP Hazardous Drug Compounding Pharmacy Technician Toolkit
- Oncology Nursing Society
- American Industrial Hygiene Association (AIHA) Hazardous Drug Surface Contamination Guidance Document
- ARL Bio Pharma Wipe Sampling Test Document
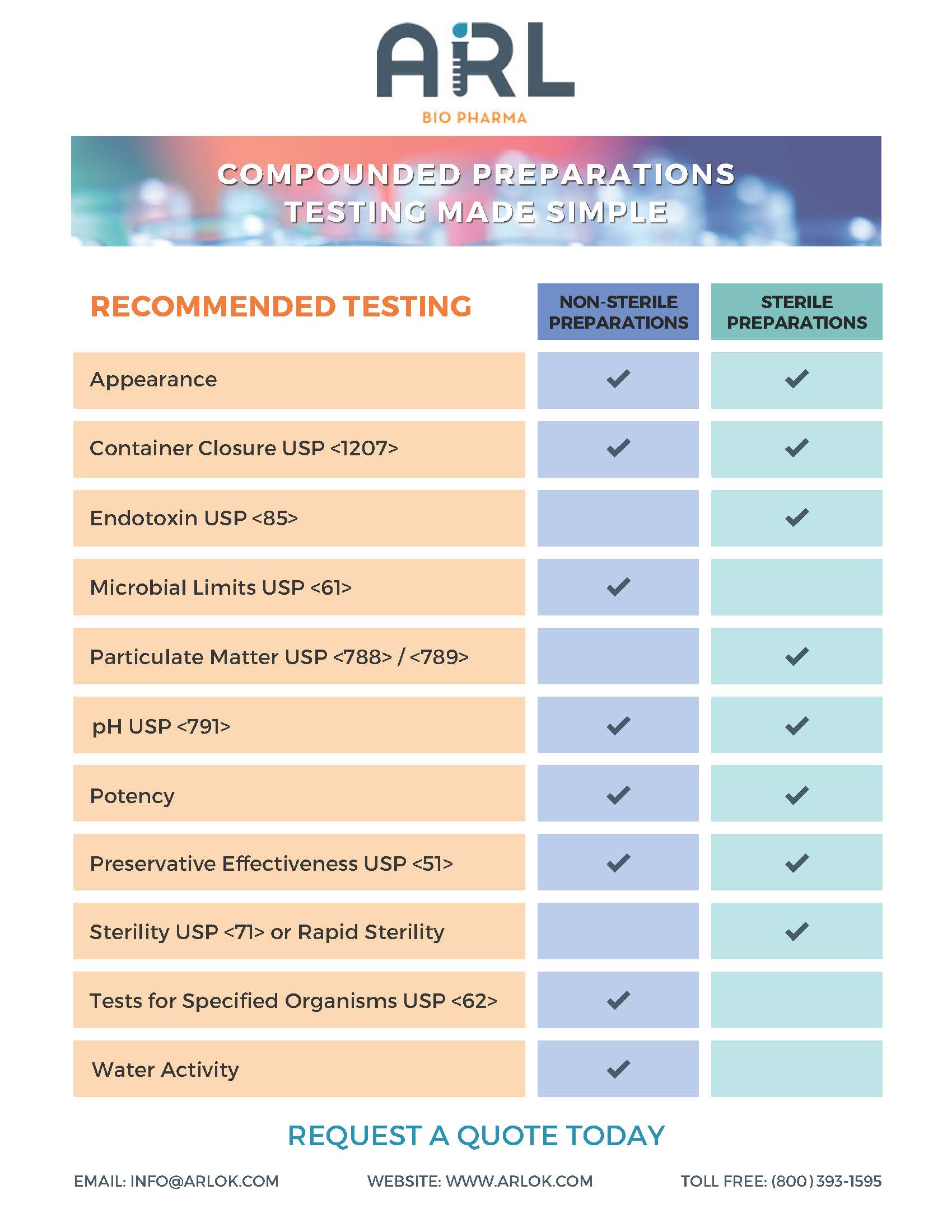
USP 797 sets minimum standards for compounded sterile preparations to prevent exposure to particulate matter that may cause patient harm.
Particulate matter is the unwanted solid material in solutions for injection that can come from different sources. It’s important pharmacies identify the source so they can implement preventive measures to manage particulate matter throughout the product lifecycle.
Particulate matter comes from three sources:
- Intrinsic stems from the internal manufacturing process. This can be introduced in the drug formulation, packaging or container, and poor cleaning systems. Examples include glass from inside containers, stainless steel from container closure systems, or rubber from the stoppers.
- Extrinsic stems from the external production environment and human error. This can be introduced during improper gowning in a cleanroom and other external behavior. Examples include clothing, hair, metal, plastic, or paint that fall into sample containers.
- Inherent stems from a product’s characteristic that may contain a small level of unintended particulate matter, even after careful formulation
CSPs must pass a visual inspection at the completion of compounding and before release and dispensing. This assures its physical appearance is free from inappropriate visible particulates, foreign matter, discoloration, or other defects. The limit of visibility depends on the test conditions and the nature of the particulate matter. Particles larger than 100 µm are generally considered visible particles.
Subvisible particles are in the range of 2-100 µm. United States Pharmacopeia has three main chapters that describe testing methods and specifications for particulate matter:
- USP <787> Subvisible Particulate Matter in Therapeutic Protein Injections
- USP <788> Particulate Matter in Injections
- USP <789> Particulate Matter in Ophthalmic Solutions
If the Category 3 CSP is an injection or an ophthalmic solution, particulate matter testing is conducted once per formulation with acceptable results. The CSP passes particulate matter testing if unintended particulates in the dosage form do not exceed USP established limits.
For parenteral dosage forms where the nature of the contents or container-closure system permits only limited inspection of the total contents, USP <1> states the 100% inspection of the lot should be supplemented with the inspection of constituted (dried) or withdrawn (from a dark amber container) contents of a sample of containers from the lot for particulate matter testing.
Pharmacies must establish processes to monitor and mitigate potential sources of particulates in final products. By doing so, pharmacies not only ensure the quality of the products but also safeguard the health and well-being of the patients.
For more information on particulate matter testing, contact ARL at 800-393-1595 or info@arlok.com.
Resources:
- USP <1> Injections and Implanted Drug Products (Parenterals)—Product Quality Tests
- USP <787> Subvisible Particulate Matter in Therapeutic Protein Injections
- USP <788> Particulate Matter in Injections
- USP <789> Particulate Matter in Ophthalmic Solutions
- USP <797> Pharmaceutical Compounding—Sterile Preparations
- USP <1787> Measurement of Subvisible Particulate Matter in Therapeutic Protein Injections
- USP <1788> Methods for the Determination of Subvisible Particulate Matter
- FDA Inspection of Injectable Products for Visible Particulates Draft Guidance for Industry
A Compounding Record (CR) is a document that records the preparation of a compounded medication. It is important to maintain compounding records to demonstrate compliance with USP standards.
For nonsterile preparations, a CR must be created for all preparations to ensure the traceability of all components. This document must be reviewed for completeness before the preparation is released. The name, or unique identifier, of the reviewer and the date must be recorded. A master formulation record (MFR), a detailed recipe for preparing a drug product, may be used as the basis for a CR by incorporating spaces to fill in the necessary information to complete the compounding record.
For sterile preparations, a CR must be created for all Category 1, Category 2, and Category 3 preparations and immediate-use CSPs prepared for more than one patient. A written or electronic template, prescription, medication order, label, retrievable electronic information stored in an Automated Compounding Device (ACD), or a copy of the MFR may serve as a compounding record.
For more information on Compounding Records and required contents, visit USP <795> and <797>.
Compounding Records and Quality Control Testing
Compounding Records are essential to provide testing laboratories with information on the drug product’s preparation. Submitting a CR with every sample helps improve the on-time delivery of test results and reduces potential out-of-specification (OOS) investigations.
ARL uses the CR to review:
- Actual quantities used to compound a preparation
- Total quantity of preparations made in a compounding process
- Balance quantities
- Purity of starting material (e.g. moisture content accounted for in compounding)
- Calculations used in compounding (e.g. salt form conversions)
Submitting CRs can help identify and resolve discrepancies during sample receiving, test analysis, and reporting of the results.
For more information about submitting Compounding Records, contact ARL at 800-393-1595 or info@arlok.com.
A Master Formulation Record (MFR) is a detailed record of procedures that describes how the drug product is to be prepared. This documentation is an important component of regulatory compliance and effective process control.
According to USP <795> and <797>, a MFR must be created for:
- Each unique formulation of a compounded nonsterile preparation (CSNP)
- Compounded sterile preparations (CSP) for more than one patient
- CSP prepared from nonsterile ingredient(s)
Any changes or alterations to the MFR must be approved and documented according to the facility’s SOPs.
Visit USP <795> and <797> for more information on a Master Formulation Record and its required contents.
Master Formulation Records and Quality Control Testing
Master Formulation Records are essential to providing testing laboratories with information on how the drug product is prepared. It gives a thorough view of your pharmacy’s processes and aids ARL in providing high-quality results.
Submitting an MFR with every sample helps improve the on-time delivery of test results to your pharmacy and reduces potential out-of-specification (OOS) investigations.
ARL uses the MFR to:
- Know which diluents and methods to use to extract API for testing
- Establish method suitability to prove the test method is suitable for its intended use
- Match unique formulations to corresponding unique formulation identification numbers
Pharmacists must update MFRs with any changes or alterations to the compounded preparation.
Changes or alterations include, but are not limited to:
- A change in preservative or preservative amount
- Example: updating preservative and/or preservative amount in response to a failing antimicrobial effectiveness test
- A change in the vehicle
- Example: vehicle supplier changes due to availability
- A change in suppliers where the new supplier’s formulation does not match the old supplier
- Example: 0.9% normal saline vs. 0.45% normal saline
- Example: salt form vs. non-salt form
- Example: preservative-free vs. preserved vehicle
- A change in the amount of active ingredients used
- Example: active ingredient updated due to a potency failure
- A change in notes
- A change in active ingredient starting material – raw material vs. triturate vs. commercial product
- A change in a lot of active ingredients received from a supplier
- Example: purity factor or water content
It is crucial to document the compounding process of each drug product by creating a Compounding Record (CR) in addition to the Master Formula Record (MFR). It is also important to record the balance quantities in a CR. The MFR can be used as a reference while creating a CR. One way to do this is by incorporating spaces in the MFR to fill in the necessary information to complete the compounding record.
Visit USP <795> and <797> for more information on a compounding record and its required contents.
Submitting MFRs and CRs can help identify and resolve discrepancies during sample receiving, test analysis, and/or reporting of the results. Thus, reducing the need for additional client communication to answer questions that might arise.
For more information about submitting Master Formulation Records, contact ARL at 800-393-1595 or info@arlok.com.
Formulation Change Example
| Original Formulation | ||
|---|---|---|
| Ingredient | Amount Used | Concentration |
| Methylcobalamin | 5 mg | 50 mg/mL |
| Benzyl Alcohol | 0.9 g | 9 mg/mL |
| Sterile Water | 100 mL | N/A |
| Change in Preservative | ||
|---|---|---|
| Ingredient | Amount Used | Concentration |
| Methylcobalamin | 5 mg | 50 mg/mL |
| Benzalkonium Chloride | 0.9 g | 9 mg/mL |
| Sterile Water | 100 mL | N/A |
| Change in Preservative Amount | ||
|---|---|---|
| Ingredient | Amount Used | Concentration |
| Methylcobalamin | 5 mg | 50 mg/mL |
| Benzyl Alcohol | 1.5 g | 15 mg/mL |
| Sterile Water | 100 mL | N/A |
| Change in Vehicle | ||
|---|---|---|
| Ingredient | Amount Used | Concentration |
| Methylcobalamin | 5 mg | 50 mg/mL |
| Benzyl Alcohol | 0.9 g | 9 mg/mL |
| Normal Saline | 100 mL | *Adds NaCl 9mg/mL |
| Addition of Excipient | ||
|---|---|---|
| Ingredient | Amount Used | Concentration |
| Methylcobalamin | 5 mg | 50 mg/mL |
| Benzyl Alcohol | 0.9 g | 9 mg/mL |
| Mannitol | 1 g | 10 mg/mL |
| Sterile Water | 100 mL | N/A |
| Change in Active Ingredient Amount | ||
|---|---|---|
| Ingredient | Amount Used | Concentration |
| Methylcobalamin | 5.4 mg | 54 mg/mL |
| Benzyl Alcohol | 0.9 g | 9 mg/mL |
| Sterile Water | 100 mL | N/A |
| Change in Total Amount Made | ||
|---|---|---|
| Ingredient | Amount Used | Concentration |
| Methylcobalamin | 10 mg | 50 mg/mL |
| Benzyl Alcohol | 1.8 g | 9 mg/mL |
| Sterile Water | 200 mL | N/A |
Antimicrobial preservatives are excipients added to aqueous pharmaceutical products.
Nonsterile dosage forms may have preservatives added to protect them from the growth of microorganisms inadvertently introduced during or after the compounding process. USP <795> Pharmaceutical Compounding—Nonsterile Preparations requires passing USP <51> Antimicrobial Effectiveness Testing data on aqueous, compounded non-sterile preparations if extending the beyond use date past the limits in USP <795> Table 4. USP defines an aqueous preparation as one with a water activity greater than 0.6.
Sterile dosage forms packaged in multiple-dose containers have preservatives added to inhibit the growth of microorganisms that may be introduced from repeatedly withdrawing individual doses. USP <797> Pharmaceutical Compounding—Sterile Preparations requires passing antimicrobial effectiveness testing data on aqueous, multiple-dose compounded sterile products.
Both <795> and <797> state antimicrobial effectiveness testing must be conducted and passed once for each formulation in accordance with USP <51> for the particular container closure system in which it will be packaged. For a product to pass antimicrobial effectiveness testing, formulations containing preservatives are expected to meet minimal efficacy standards, whether packaged as multidose or unit doses. The compounder may rely on results from an FDA-registered facility or published in peer-reviewed literature sources if the formulation (including any preservative) and container closure materials of composition are the same as those tested (unless a bracketing study is performed). When a bracketing study is performed, antimicrobial effectiveness testing may be performed on a low concentration and on a high concentration of the active ingredient in the formulation to establish preservative effectiveness across various strengths of the same formulation (e.g., bracketing). The concentration of all other ingredients (including preservatives) must fall within the bracketed range.
FDA requires 503B Outsourcing Facilities to have passing antimicrobial effectiveness testing data on sterile drug products labeled as multiple-dose and for aqueous non-sterile products labeled as multiple-dose. If antimicrobial effectiveness testing was previously performed using the subject formulation and container-closure system, preservative content testing may be used in lieu of a full antimicrobial effectiveness study. Appropriate specifications for aqueous drug products labeled as multiple-dose include assurances that the product is adequately self-preserving or contains appropriate preservative content to limit microbial proliferation of microorganisms and assures the product maintains its quality and purity for each dose. Antimicrobial effectiveness studies only need to be conducted once for each formulation and container-closure system, and a bracketing or matrixing approach can be considered to minimize the amount of testing.
Testing of Products
USP <51> is designed to challenge the antimicrobial properties of the drug product by adding large amounts of microorganisms into the preserved compounded preparation to simulate a contamination event during or after compounding. The mixture of microorganisms and preserved drug product are incubated and the number of colony forming units (CFUs) are counted over time.
The added microorganisms include:
- Candida albicans
- Aspergillus brasiliensis
- Escherichia coli
- Pseudomonas aeruginosa
- Staphylococcus aureus
The time points at which the CFUs are counted depends on which of the 4 USP <51> defined categories applies to the product being tested. The goal of this periodic plating is to demonstrate the product’s ability to control microbial growth over time.
USP <51> Table 1. Compendial Product Categories
| Categories | Product Description |
|---|---|
| 1 | Injections; other parenterals including emulsions, otic products, sterile nasal products, and ophthalmic products made with aqueous bases or vehicles |
| 2 | Topically used products made with aqueous bases or vehicles; nonsterile nasal products and emulsions, including those applied to mucous membranes |
| 3 | Oral products other than antacids, made with aqueous bases or vehicles |
| 4 | Antacids made with an aqueous base |
The acceptance criteria to determine if the drug product is sufficiently preserved is also defined in USP <51> based upon the drug category and the type of microorganism.
USP <51> Table 3. Criteria for Tested Microorganisms
| For Category 1 Products | |
|---|---|
| Bacteria | NLT 1.0 log reduction from the initial calculated count at 7 days, NLT 3.0 log reduction from the initial count at 14 days, and no increase from the 14 days’ count at 28 days |
| Yeast & Molds | No increase from the initial calculated count at 7, 14, and 28 days |
| For Category 2 Products | |
| Bacteria | NLT 2.0 log reduction from the initial count at 14 days, and no increase from the 14 days’ count at 28 days |
| Yeast & Molds | No increase from the initial calculated count at 14 and 28 days |
| For Category 3 Products | |
| Bacteria | NLT 1.0 log reduction from the initial count at 14 days, and no increase from the 14 days’ count at 28 days |
| Yeast & Molds | No increase from the initial calculated count at 14 and 28 days |
| For Category 4 Products | |
| Bacteria, Yeast, and Molds | No increase from the initial calculated count at 14 and 28 days |
To obtain a passing result, all criteria listed in Table 3 must be met for the applicable drug product category.
For more information on Antimicrobial Effectiveness Testing:
- USP <51> Antimicrobial Effectiveness Testing
- USP <795> Pharmaceutical Compounding Non-Sterile Preparations
- USP <797> Pharmaceutical Compounding Sterile Preparations
- FDA Current Good Manufacturing Practice—Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act
Contact ARL to request a quote info@arlok.com.
James Zellner, ARL Bio Pharma Technical Sales
In the upcoming version of USP <797> Pharmaceutical Compounding – Sterile Preparations, the method of sterilization has a significant impact on the maximum allowable Beyond-Use date (BUD) a compounded preparation may be assigned. The two approaches to sterilization in the chapter are aseptic processing and terminal sterilization. Terminal sterilization provides the longest BUDs possible but may not be compatible with the compounded preparation due to the conditions required to sterilize. Ultimately, the appropriate sterilization method for a particular formulation depends on the product, with the allowable BUD a consequence.
The new version of USP <797> equates the maximum allowable BUD with the method of sterilization. USP defines terminal sterilization as the “application of a lethal process (e.g., steam, dray heat, irradiation) to sealed containers for the purpose of achieving a predetermined PNSU (Probability of Non-Sterile Unit) of greater than 10-6 or a probability of less than one in a million of a nonsterile unit”. Aseptic processing is defined as “a method by which separate, sterile components are brought together under conditions that maintain their sterility. The components can either be purchased as sterile or, when starting with nonsterile components, can be separately sterilized prior to combining.” Tables in the chapter are presented for Category 2 and Category 3 products that state the conditions that must be met to assign a particular BUD.
Table 1. BUD Limits for Category 2 CSPs – Assuming a Sterility Test has Been Performed and Passed
| Compounding Method | Controlled Room Temp | Refrigerated | Frozen |
|---|---|---|---|
| Aseptically Processed | 30 Days | 45 Days | 60 Days |
| Terminally Sterilized | 45 Days | 60 Days | 90 Days |
Table 2. BUD Limits for Category 3 CSPs – Assuming a Sterility Test has Been Performed and Passed
| Compounding Method | Controlled Room Temp | Refrigerated | Frozen |
|---|---|---|---|
| Aseptically Processed | 60 Days | 90 Days | 120 Days |
| Terminally Sterilized | 90 Days | 120 Days | 180 Days |
Based on the tables above, the maximum BUD allowed in USP <797> is 180 days if the product is terminally sterilized and stored frozen. BUDs are reduced from this maximum based on the storage conditions and sterilization method. Terminal sterilization is given a preferential BUD since a sterility assurance level can be reliably calculated and the variable risk of contamination during compounding is eliminated since a sealed, finished product is sterilized at the very end of the process. Of note, USP does not consider filter sterilization a Terminal Sterilization method. Aseptic processing, without a final, sealed container sterilization step, requires aseptic conditions be maintained throughout the process to deliver a sterile finished product. When choosing or changing a sterilization method for a preparation, a BUD study utilizing a validated, stability indicating method is necessary. If BUD data was previously demonstrated using aseptic processing, and the pharmacy wants to now utilize terminal sterilization, the BUD study must be repeated to demonstrate the product meets all test criteria.
The tolerance of the product, in conjunction with the tolerance of the container system, dictate whether a particular formulation may be terminally sterilized. Temperature-sensitive preparations may be impossible to sterilize terminally, given the typical temperatures and times required (121o C for 15 minutes or more). Container systems may degrade under terminal sterilization conditions. When considering the use of terminal sterilization, the whole formulation must be taken into account. Even if the active pharmaceutical ingredient is stable, excipients, to include any preservatives, may break down, rendering them inert for the intended purpose.
If terminally sterilized, USP requires those products made from nonsterile ingredients, or compounded using nonsterile devices, be sterilized within 6 hours of compounding. This is to minimize the generation of bacterial endotoxins. Logistical and scheduling issues may occur depending on the number of product lots made, container sizes, and throughput of the terminal sterilization process. USP also requires terminal sterilization specific measures be taken for SOPs and validation of the sterilization cycle. SOPs specifying the temperature, duration, load conditions, and biological indicator use must be written. Training and competency of personal on the terminal sterilization process must be recorded and kept. Utilizing terminal sterilization requires an in- depth knowledge of the product, logistical considerations, thorough SOPs, and continuous monitoring of the effectiveness of the procedure to properly implement.
A compounded sterile preparation’s BUD is derived from numerous factors. One of the most important being the method used to achieve a sterile finished product. Terminal sterilization and aseptic processing both have advantages and disadvantages which must be thoroughly examined to decide which is best for each product.
References:
- USP <797>