What is particulate matter and why is testing performed?
Particulate matter is unwanted solid material in solutions for injection. Particle counts that exceed USP limits in intravenous drug products could block blood flow inside the patient’s body causing paralysis or death.
Particulate matter testing is a good indicator of the quality of a manufacturing process and the containers used. If the containers are not particle free on the inside, this will introduce particulates into the drug product. If the instrumentation used is not clean, unwanted solid matter could end up in a final drug product and cause patient harm.
Where does particulate matter come from?
Particulate matter comes from three sources:
- Intrinsic stems from the internal manufacturing process. This can be introduced in the drug formulation, packaging or container, and poor cleaning systems. Examples include glass from inside containers, stainless steel from container closure systems, or rubber from the stoppers.
- Extrinsic stems from the external production environment and human error. This can be introduced during improper gowning in a cleanroom and other external behavior. Examples include clothing, hair, metal, plastic, or paint that fall into sample containers.
- Inherent stems from a product’s characteristic that may contain a small level of unintended particulate matter, even after careful formulation
How is particulate matter tested?
United States Pharmacopeia has three main chapters that describe testing methods and specifications for particulate matter:
- USP <787> Subvisible Particulate Matter in Therapeutic Protein Injections
- USP <788> Particulate Matter in Injections
- USP <789> Particulate Matter in Ophthalmic Solutions
USP also has chapters that give broader information and relate to the test methodology:
- USP <771> Ophthalmic Products – Quality Tests
- USP <1788> Methods for the Determination of Subvisible Particulate Matter
- USP <1788.1> Light Obscuration Method for the Determination of Subvisible Particulate Matter
- USP <1788.2> Membrane Microscope Method for the Determination of Subvisible Particulate Matter
ARL Bio Pharma uses validated methodology to test particulate matter samples. Our laboratory uses two methods to test for particulate matter: Method 1 and Method 2. There are advantages and disadvantages to each method. ARL determines which method to use based on sample type.
Method 1 uses a light obscuration particle counter to test the sample. A probe goes into the sample container. The sample travels up through the laser’s path and particles are counted. When testing is complete, the sample is discarded out of the system to a waste container
Method 2 uses a microscope, and an analyst counts the particles. The particles present in a sample are collected on a membrane filter and then viewed under a microscope for counting. The analyst uses a graticule inside the microscope lens to determine the size and count of the particles based on USP criteria.
| Method 1 | Method 2 | |
|---|---|---|
| Appearance |
|
|
| Appearance |
|
|
What are the USP Particle Size Limits for Light Obscuration and Microscopy?
USP <787> Subvisible Particulate Matter in Therapeutic Protein Injections
| Sample Volume | ≥10 µm | ≥25 µm |
|---|---|---|
| ≤100 mL | 6000 per container | 600 per container |
| 100 mL | 25 per mL AND 6000 per container | 3 per mL AND 600 per container |
USP <788> Particulate Matter in Injections
| Method | Sample Volume | ≥10 µm | ≥25 µm |
|---|---|---|---|
| 1-a | >100 mL | NMT 25 particles/mL | NMT 3 particles/mL |
| 1-b | ≤100 mL | NMT 6000 particles/container | NMT 6000 particles/container |
| 2-a | >100 mL | NMT 12 particles/mL | NMT 2 particles/mL |
| 2-b | ≤100 mL | NMT 3000 particles/container | NMT 300 particles/container |
USP <789> Particulate Matter in Ophthalmic Solutions
| Method | ≥10 µm | ≥25 µm | ≥50 µm |
|---|---|---|---|
| 1 | NMT 50 particles/mL | NMT 5 particles/mL | NMT 2 particles/mL |
| 2 | NMT 50 particles/mL | NMT 5 particles/mL | NMT 2 particles/mL |
For more information on particulate matter testing, contact (800) 393-1595 or info@arlok.com
Reviewing data is an important part of an OOS investigation. Raw data review includes:
Laboratory Calculations
ARL’s data reviewer checks all the calculations recorded in the laboratory notebooks and ensures the arithmetic is accurate. The laboratory examines water content and molecular weight calculations for transcription errors of active pharmaceutical ingredient standards.
Chromatograms and Spectra
Our laboratory also reviews the chromatograms and spectra to ensure the data passes quality requirements:
- peak shape (width and symmetry)
- peak height
- proper elution
The data reviewer evaluates the retention time (RT) to confirm the sample API peak comes out at the same time as the standard API peak in the chromatogram.
Standards and Reagents
During review, ARL verifies the correct standard was used. It is important correct standards are used as different forms of APIs may have different retention times. ARL uses USP grade material as the standard for non-cGMP work and USP reference standards for cGMP work, if available. The data reviewer makes sure the standard did not contribute to the out of specification result.
We also ensure the reference standard was stored properly. If standards are kept in abnormal storage conditions, results can trend high as the standard degrades.
Any other required procedures such as evaluating water content or drying was conducted properly on the reference material are reviewed during OOS investigations.
Trends
Trends can occur at the instrumentation, sample, or analyst level.
An instrumentation failure occurs when an instrument is giving bias results. ARL’s metrology department ensures all laboratory instruments are calibrated and comply with method requirements. If an instrument is not regularly serviced, API residue can build up in the instrument’s loop and cause high results.
Sample trends refer to getting results either always low or always high. If a sample is resulting low, our laboratory reviews the extraction procedure and chemist trends. If a sample is always high, our laboratory reviews the standard. Data reviewers also look at client sample trends to see if the preparation has a history of out of specification. In stability studies, results should follow a high-to-low trend. Low-to-high trends may indicate sample preparation or vehicle/matrix issues. Evaporation in some formulations can also cause low to high trends due to specific excipients and/or container integrity.
After a raw data review, our analysts perform visual inspections to see if there is a visible reason a sample may be out of specification due to particulate matter or binding of API within the vehicle. Our analysts may also contact clients before re-testing as sometimes samples are sent for testing because the client suspects the sample may be OOS.
ARL’s out-of-specification investigations use a hybrid approach to review data, formulations, and tests. Our data reviewers consult with ARL’s scientific directors and clients to ensure we provide a 360-view investigation and evaluate all aspects of the laboratory, data, and client and analyst trends. Typical investigations are concluded within one week or within 4 business days after the due date.
For more information on OOS investigations, contact ARL 800-393-1595 or info@arlok.com
Look back at OOS Investigations Part 1 – Sample Information and Preparation
United States Pharmacopeia monographs establish standards for identity, strength, quality, and purity of medicines. Monographs list tests and acceptance criteria (usually expressed in percentage ranges or limits). Drug substances and products should meet the acceptance criteria to be considered acceptable for its intended use. An Out of Specification (OOS) occurs when a test fails to meet acceptance criteria. An OOS Investigation follows an OOS test result and is performed to determine if there is a root cause. In this article we will look specifically at potential causes of potency OOS results of finished drug products. Understanding the factors that contributed to the potency failure allows the pharmacist and laboratory to define a strategic approach in eliminating future failures. The more specific the findings, the easier it is to properly address the cause.
If an OOS result occurs, the Food and Drug Administration requires:
- a valid reason must be determined to invalidate the OOS results (for it to be discarded)
- investigation into the cause of the OOS result must be conducted, and
- that the results from additional tests should not be averaged unless there is scientifically sound reason that results should be averaged.
The first steps of ARL Bio Pharma’s OOS investigation are to review the formulation sheet and test procedures.
Common causes of potency OOS results include:
Content Uniformity
Here we are specifically looking into how much drug is contained in individual dosage units such as tablets and capsules. Variation in the amount of drug in a capsule can come from many steps during the process (mixing and filling).
This is an example of potency data from individual dosage units without uniform drug content:
- Test result from capsule 1: 88%
- Test result from capsule 2: 92%
- Test result from capsule 3: 115%
The laboratory cannot average Test results 1, 2, and 3, because the data points to a content uniformity issue with the individual dosage units. According to FDA, all test results must appear on the Certificate of Analysis (COA) unless there is a clear, scientific reason the initial test result is invalid. To overcome non-uniform samples, ARL combines multiple individual dosage units into a single test sample. Even with this practice, pharmacies should understand if they are producing uniform drug products.
Recommendation:
- Test individual units from beginning, middle, and end to determine if uniformity is a root cause
- Properly blend drug with excipients
- Ensure uniform amount of blend is added to each dosage unit
Overfill: a commercial product is added to a pre-filled IV bag
If pharmacies do not account for overfill, potency test results are likely to be low. ARL offers a fill volume test to take a measurement of the solution in an IV bag. This measurement is used to calculate the final potency results.
Example: A pharmacy may begin with an IV bag labeled as containing 100mL, but the actual volume is more than 100mL. If the pharmacy calculates the amount of commercial product based on the IV bag labeled volume instead of the actual volume, the potency test results will be lower than expected and potentially OOS.
Recommendation:
- Check fill volume and account for the extra volume during compounding.
- Conduct a fill volume test to ensure correct solution measurement on potency results.
- Label the finished drug product with the amount of drug per bag instead of as a concentration.
Correct salt form or hydrate
If pharmacies do not account for salt forms, potency results will vary due to incorrect formulation calculations and test result reporting.
Example: Sildenafil vs. Sildenafil Citrate
If a pharmacy requests potency testing of the incorrect form of Sildenafil, the potency result will be off by at least 28% due to the molecular weight difference between these two forms of Sildenafil:
- Sildenafil Molecular Weight – 474.58
- Sildenafil Citrate Molecular Weight – 666.70
Recommendation:
- Check starting material certificate of analysis to verify the drug form
- Check the compounding formulation sheet to make sure the calculations are based on the drug form being used
- Always submit a formulation sheet with potency test requests
If a pharmacy does not account for the hydrate form, or water, in an API (active pharmaceutical ingredient), the potency test will give low results. Every API has some form of water.
Example: T3/T4 may have as much as 8% water. If water is not accounted for, the potency results will be sub-potent.
Recommendation:
- Check water content on API before compounding. Remember that some APIs will absorb more water overtime. The more water absorbed, the lower the potency results will be.
Solubility issues
Here we are talking specifically about test sample preparation in the testing laboratory. Most compounded drug products require drug extraction and dilution procedures before testing. Sometimes multiple diluents (substances used to extract and dilute) are utilized.
Example:
A Bupivacaine HCl topical cream may need isopropyl alcohol to break down the cream portion, and then water to dissolve the API into solution for testing. However, these diluents may not be a suitable mix for the HPLC column, mobile phase, and even wavelength used to determine the results.
ARL Bio Pharma has over 22 years of testing experience to estimate the proper API extraction procedures. However, with the large variety of formulation tested, proper extraction is evaluated during the OOS investigation as a potential cause of the OOS result.
Spills
This refers to sample spills within the laboratory.
Example:
If liquid was spilled from a suspension sample before taking the sample for potency testing, the lab could not guarantee accurate results. Similarly, if a titration powder is spilled and the lab preps from the remainder, the chemist has not taken a representative sample.
ARL Bio Pharma contacts the client to determine how to proceed if a spill or leak has occurred. If a sample is spilled, the laboratory documents in the notebook and requests additional sample from the client.
Inaccurate volumetric transfer
This may occur if the incorrect volumetric flask is used when preparing a sample for HPLC testing.
Example:
A 200mL and a 250mL flask look very similar. Using the wrong flask can lead to inaccurate potency results.
ARL Bio Pharma looks for inaccurate volumetric transfers during the OOS investigation data review to ensure appropriate laboratory supplies are used.
Inaccurate weight
This refers to the weight of a cream/lotion/ointment/gel sample expressed as weight/volume.
Example:
Formulation sheets help identify the theoretical density of a solid sample matrix expressed as weight/volume. If the theoretical density is around one, but the chemist records a value significantly lower than the theoretical weight per 1ml, then re-tests are performed by a different chemist.
- Chemist 1 may weigh 0.9 grams for 1mL of sample as recorded on the notebook page.
- Chemist 2 retests the sample and weighs 0.98, two times, for 1 mL sample as recorded on the notebook page.
The information recorded on the notebook pages may indicate a problem with transferring of the material in the first test, which is usually caused by air pockets in the sample. If there are air pockets in the measured 1ml volume, then the true volume is not 1ml.
ARL Bio Pharma chemists are trained to transfer the sample without incorporating air into the sample.
Next month, we will examine OOS Investigations Part 2 and review raw data, standards and reagents, and trends.
Look forward to OOS Investigations Part 2 – Raw Data, Standards and Trends
Minor Chapters, Major Impacts – What United States Pharmacopeia Chapters <51>, <61>, <62>, and <1207> mean for your compounding practice
Brian Kelley, BS, ARL Bio Pharma Director of Business Development
International Journal of Pharmaceutical Compounding, Vol. 25, No. 2, March / April 2021, p. 115-124
Abstract
The United States Pharmacopeial Convention, Inc. recommends within the standards of the United States Pharmacopeia that compounding pharmacies have staff dedicated to quality assurance and quality control to ensure patients are receiving safe medications. The quality-control program must include testing. While compounding pharmacies have grown familiar with potency, sterility, and endotoxin testing, there are many more tests recommended within the United States Pharmacopeia that are critical for evaluating the quality of compounded preparations. This article discusses when a few of these tests should be utilized, how to assign acceptance criteria, and how test results are obtained.
Quality Assurance and Quality Control
United States Pharmacopeia (USP) chapters <795> and <797> state that compounding pharmacies should have staff dedicated to quality assurance (QA) and quality control (QC). QA is responsible for procedures, activities, and oversight, while QC is responsible for sampling, testing, and documenting the test results to ensure that specifications have been met before dispensing compounded preparations. USP Chapter <1163> Quality Assurance in Pharmaceutical Compounding is referred to in chapters <795> and <797> because it provides guidance on how to set up QA/QC programs in a compounding pharmacy and makes recommendations for appropriate tests to conduct on compounded preparations. TESTING The goal of testing compounded preparations is to determine the adequacy of the compounding process and the quality of the preparation. Not only is testing the finished preparation recommended in USP Chapter <1163>, but so is testing of intermediates or stock solutions. Testing of stocks is important because the final preparation is likely to be out of specification (OOS) if the stock is not correct. Chapter <1163> also says that compounders must have a basic understanding of pharmaceutical analysis and that test acceptance criteria must be determined prior to testing. The Chapter acknowledges that testing of all preparations is not practical or required, but compounders should know the importance of:
- testing,
- when to test,
- what to test,
- the appropriate test method and equipment,
- how to interpret test results,
- limits of the test, and
- actions to take when a test result is OOS.
Chapter <1163> lists many tests and provides details on the appropriateness of each test. This article focuses on three tests in USP Chapter <1163> and one from the U.S. Food and Drug Administration (FDA) that are not well-known but are important for checking the quality of compounded nonsterile and sterile preparations for release and beyond-use dating (BUD). The tests listed in Chapter <1163> and discussed in this article relate to chapters:
- <61> Microbial Enumeration Tests (applies to non-sterile products)
- <62> Tests for Specified Organisms (a recommended test on nonsterile products)
- <51> Antimicrobial Effectiveness Testing (applies to preserved multi-dose nonsterile and sterile aqueous products)
- <1207> Package Integrity Testing – Sterile Products (applies to sterile and nonsterile preparations)
| TABLE 1 OF USP CHAPTER <1111> | |||
|---|---|---|---|
| UNITED STATES PHARMACOPEIA CHAPTER <1111> TABLE 1. ACCEPTANCE CRITERIA FOR MICROBIOLOGICAL QUALITY OF NONSTERILE DOSAGE FORMS | |||
| ROUTE OF ADMINISTRATION | TOTAL AEROBIC MICROBIAL COUNT (CFU/g OR CFU/mL) | TOTAL COMBINED YEASTS/MOLDS COUNT (CFU/g OR CFU/mL) | SPECIFIED MICROORGANISM(S) |
| Nonaqueous preparations for oral use | 103 | 102 | Absence of Escherichia coli (1 g or 1 mL) |
| Aqueous preparations for oral use | 102 | 101 | Absence of Escherichia coli (1 g or 1 mL) |
| Rectal use | 103 | 102 | — |
| Oromucosal use | 102 | 101 |
Absence of Staphylococcus aureus (1 g or 1 mL) Absence of Pseudomonas aeruginosa (1 g or 1 mL) |
| Gingival use | 102 | 101 |
Absence of Staphylococcus aureus (1 g or 1 mL) Absence of Pseudomonas aeruginosa (1 g or 1 mL) |
| Cutaneous use | 102 | 101 |
Absence of Staphylococcus aureus (1 g or 1 mL) Absence of Pseudomonas aeruginosa (1 g or 1 mL) |
| Nasal use | 102 | 101 |
Absence of Staphylococcus aureus (1 g or 1 mL) Absence of Pseudomonas aeruginosa (1 g or 1 mL) |
| Auricular use | 102 | 101 |
Absence of Staphylococcus aureus (1 g or 1 mL) Absence of Pseudomonas aeruginosa (1 g or 1 mL) |
| Vaginal use | 102 | 101 |
Absence of Pseudomonas aeruginosa (1 g or 1 mL) Absence of Staphylococcus aureus (1 g or 1 mL) Absence of Candida albicans (1 g or 1 mL) |
| Transdermal patches (limits for one patch including adhesive layer and backing) | 102 | 101 |
Absence of Staphylococcus aureus (1 patch) Absence of Pseudomonas aeruginosa (1 patch) |
| Inhalation use (special requirements apply to liquid preparations for nebulization) | 102 | 101 |
Absence of Staphylococcus aureus (1 g or 1 mL) Absence of Pseudomonas aeruginosa (1 g or 1 mL) Absence of bile-tolerant Gram-negative bacteria (1 g or 1 mL) |
| cfu = colony-forming agents Source: United States Pharmacopeial Convention, Inc. United States Pharmacopeia–National Formulary. Rockville, MD: United States Pharmacopeial Convention, Inc.; Current Edition. | |||
United States Pharmacopeia Chapter <61> Microbial Enumeration Tests
USP <61> applies to nonsterile products, which includes finished nonsterile compounded preparations, and may be used to assess intermediates of sterile compounds. This is a test that determines how many microorganisms are present in nonsterile drug products and sterile products that are made using nonsterile components prior to sterilization of the final preparation. The microbial enumeration test is often referred to as “bioburden” or “microbial limits.” In 483s issued to 503A pharmacies, the FDA has also referred to these tests as “yeast and mold counts” and “presence of microorganisms.” As stated in Chapter <1163>, the acceptance criteria must be defined prior to test initiation. Acceptance criteria are not provided in Chapter <61>. Instead, acceptance criteria recommendations are provided in USP Chapter <1111> or the USP monograph. The limits listed in Table 1 of USP Chapter <1111> list how many microorganisms can be present in nonsterile compounded preparations. Separate criteria are provided for each of the two tests included in USP Chapter <61>.
| TABLE 2 OF USP CHAPTER <1111> | ||
|---|---|---|
| UNITED STATES PHARMACOPEIA CHAPTER <1111> TABLE 2. ACCEPTANCE CRITERIA FOR MICROBIOLOGICAL QUALITY OF NONSTERILE SUBSTANCES FOR PHARMACEUTICAL USE | ||
| TOTAL AEROBIC MICROBIAL COUNT (CFU/g OR CFU/mL) | TOTAL COMBINED YEASTS/MOLDS COUNT (CFU/g OR CFU/mL) | |
| Substances for pharmaceutical use | 103 | 102 |
| Source: United States Pharmacopeial Convention, Inc. United States Pharmacopeia–National Formulary. Rockville, MD: United States Pharmacopeial Convention, Inc.; Current Edition. | ||
Table 2 of USP Chapter <1111> provides recommended limits for nonsterile components used to produce sterile preparations. USP Chapter <61> states the compounder must also consider the acceptance criteria of a nonsterile product or component when considering its use, to include:
- the nature of a nonsterile product,
- the method of application,
- the patient,
- the use of immunosuppressive agents/corticosteroids, and
- the presence of disease, wounds, and organ damage.
USP Chapter <61> testing is performed by plating the prepared sample onto two types of growth media. The sample preparation details are determined during method suitability. The growth medias and sample mixtures are incubated at defined temperatures and durations. After the incubation period, the colony numbers are counted, and the results are calculated to correct for any dilution of the compounded preparation that occurred during test-sample preparation. The test results are then compared to the defined acceptance criteria to determine if the nonsterile preparation passes or fails the test. The goal of USP Chapter <61> testing is to ensure that there is low bioburden in nonsterile compounded preparations and intermediates.
United States Pharmacopeia Chapter <62> Tests For Specified Organisms
While USP Chapter <61> demonstrates the total number of microorganisms, USP Chapter <62> tests for the presence of specific organisms. The FDA refers to this in 483s as tests for “objectionable organisms” or lists “specific organisms” that cannot be present in nonsterile preparations. For example, the FDA has stated “freedom from Pseudomonas aeruginosa” or “absence of bile-tolerant Gram-negative bacteria” when referring to lack of Chapter <62> testing. To determine which organisms listed in USP Chapter <62> should not be present in nonsterile preparations, you should consider the route of administration and USP Chapter <1111>. These tests are performed similarly to USP Chapter <61> but utilize growth media designed to promote the growth of the specific target microorganism and inhibit the growth of others. Method suitability is required to determine the appropriate sample preparation steps necessary to overcome any antimicrobial properties of the compounded preparation. This ensures that the test will detect the objectionable microorganism if present. At the conclusion of the incubation, a result of “pass” or “fail” is generated. A passing result means that the target organism was not detected in the sample. A failing result means the objectionable microorganism was found. The goal of USP Chapter <62> testing is to ensure that nonsterile compounded preparations do not contain specific microorganisms of concern.
United States Pharmacopeia Chapter <51> Antimicrobial Effectiveness Testing
Many nonsterile and sterile compounded preparations contain antimicrobial preservatives to prevent the growth of microbial contamination throughout the BUD. The preservatives are needed because end users may introduce microorganisms into their compounded medication. Table 1 of this article lists common pharmaceutical preservatives, the type of formulation that is used, the effective concentration, optimal pH, and the types of organism the preservative is effective against. Examples of when microorganisms can be introduced into the compounded preparation are 1) when a patient puts unclean hands into their topical cream or 2) by repeated use of an oral product. Entry into a vial without proper aseptic technique can introduce microorganisms into sterile preparations. The test procedure described in USP Chapter <51> simulates these contamination events by adding high concentrations of 5 microorganisms to the compounded preparation. The effectiveness of the antimicrobial preservative is determined by counting the number of microorganisms present for 28 days following the contamination event. The counts occur similarly to USP Chapter <61> where the test sample is prepared, inoculated onto growth media, incubated, microorganisms counted, and results calculated to correct for any dilution of the compounded preparation while making the sample for counts. The sample preparation procedure for counting is determined during method suitability. The acceptance criteria for USP Chapter <51> vary based on the type of product and the type of organism. Table 1 in USP Chapter <51> divides the types of product into 4 categories based on the type of product and route of administration. Table 3 of USP Chapter <51> provides the acceptance criteria for each product category. Notice that each product category has 2 acceptance criteria. One is for bacteria, and the other is for yeast and molds. The goal of USP Chapter <51> testing is to prove that antimicrobial preservatives kill introduced microorganisms or prevent their proliferation.
| TABLE 1 OF USP CHAPTER <51> | |
|---|---|
| UNITED STATES PHARMACOPEIA CHAPTER <51> TABLE 1. COMPENDIAL PRODUCT CATEGORIES | |
| CATEGORY | PRODUCT DESCRIPTION |
| 1 | Injections; other parenterals including emulsions, otic products, sterile nasal products, and ophthalmic products made with aqueous bases or vehicles |
| 2 | Topically used products made with aqueous bases or vehicles; nonsterile nasal products and emulsions, including those applied to mucous membranes |
| 3 | Oral products other than antacids, made with aqueous bases or vehicles |
| 4 | Antacids made with an aqueous base |
| Source: United States Pharmacopeial Convention, Inc. United States Pharmacopeia–National Formulary. Rockville, MD: United States Pharmacopeial Convention, Inc.; Current Edition. | |
| TABLE 3 OF USP CHAPTER <51> | |
|---|---|
| UNITED STATES PHARMACOPEIA CHAPTER <51> TABLE 3. CRITERIA FOR TESTED MICROORGANISMS | |
| FOR CATEGORY 1 PRODUCTS | |
| Bacteria | NLT 1.0 log reduction from the initial calculated count at 7 days, NLT 3.0 log reduction from the initial count at 14 days, and no increase from the 14 days’ count at 28 days |
| Yeast and molds | No increase from the initial calculated count at 7, 14, and 28 days |
| FOR CATEGORY 2 PRODUCTS | |
| Bacteria | NLT 2.0 log reduction from the initial count at 14 days, and no increase from the 14 days’ count at 28 days |
| Yeast and molds | No increase from the initial calculated count at 14 and 28 days |
| FOR CATEGORY 3 PRODUCTS | |
| Bacteria | NLT 1.0 log reduction from the initial count at 14 days, and no increase from the 14 days’ count at 28 days |
| Yeast and molds | No increase from the initial calculated count at 14 and 28 days |
| FOR CATEGORY 4 PRODUCTS | |
| Bacteria, yeast, and molds | No increase from the initial calculated count at 14 and 28 days |
| NLT = not less than Source: United States Pharmacopeial Convention, Inc. United States Pharmacopeia–National Formulary. Rockville, MD: United States Pharmacopeial Convention, Inc.; Current Edition. | |
United States Pharmacopeia Chapter <1207> Package Integrity Evaluation – Sterile Products
USP Chapter <1207> describes the tests that are used to demonstrate that container-closure systems can maintain a sterile environment throughout the BUD. The FDA says testing container-closure integrity is more likely to detect problems than sterility testing throughout the storage period. USP Chapter <1207> also discusses the potential for drug product components to escape from the container-closure system and the product quality risks posed by leaks. Escaping drug product components can be problematic for both sterile and nonsterile compounded preparations. For example, if benzyl alcohol in a preserved preparation escapes from the packaging system, the concentration may fall below its effective concentration range. Other components, such as water, could also escape, increasing the compounded preparation’s drug potency. Evaluating container-closure integrity is, therefore, important for both sterile and nonsterile compounded preparations. Many container-closure integrity tests are described in Chapter <1207>. The goal of USP <1207> testing is to ensure that the product’s package provides a level of protection required to meet physicochemical label-claim specifications and maintain product sterility until time of use. All the tests are designed to challenge the entire packaging system, not the individual components. Individual components are qualified by tests described in other USP chapters. USP Chapter <1207> divides the various tests into two categories: 1) deterministic and 2) probabilistic.
Deterministic Tests
Deterministic methods are described as quantitative and definitive and preferred by the USP. Vacuum decay is a common deterministic method. Vacuum decay is performed by placing the entire packaging system into a test chamber and drawing a vacuum on the chamber. The instrument then monitors for any changes in the vacuum level pulled on the chamber. Vacuum levels are monitored by the test instrument. Vacuum level changes indicate a leak in the packaging system. Because the test results are obtained from the instrument and not by an interpretation from the test operator, the USP prefers deterministic container-closure test methods.
Probabilistic Tests
Probabilistic methods are those that are qualitative and use human judgement. The USP considers these tests less desirable than deterministic methods. Dye ingress is a common probabilistic method. A common way to perform dye ingress testing is by submerging the container-closure system inside a chamber. The chamber is then filled with a dye and a vacuum pulled. The vacuum is then turned off and the chamber allowed to return to atmospheric pressures. Most of the time, the test articles are then visually examined for the presence of dye. The differences in color perception from one analyst to the next add variability to the test results. To overcome this variability, the drug product can be tested using an analytical instrument to test for the presence of dye. However, some drug products can oxidize the dye so even a high-performance liquid chromatogram with mass spectrometry would not detect the dye. Testing for container-closure integrity is important to determine if there is a leak in the packaging system that allows microorganisms to enter or if drug preparation components can escape. The USP considers probabilistic test methods, such as dye ingress, less desirable than deterministic test methods because analyst interpretation is often required to obtain the test results.
Conclusion
The USP has many resources for compounding pharmacists to ensure their patients receive quality compounded preparations. Compounders understand that USP Chapter <795> and USP Chapter <797> provide guidance on how to make quality preparations. Both chapters state that the compounding staff must be knowledgeable of the standards in USP Chapter <795> and/or USP Chapter <797> and must also be familiar with USP Chapter <1163>. This chapter provides information on QA and QC responsibilities within a compounding pharmacy. Part of QC’s responsibility is to execute the testing program setup by the QA group. USP Chapter <1163> also provides recommendations on which tests to perform on different types of compounded preparations. Compounding is heavily regulated so compounding personnel should utilize as many tools as possible to educate themselves. Testing itself is not intended to be the QA/QC program. Testing is a lagging indicator that all the things QA is doing are working appropriately. Without testing, patient complaint or harm may be the only feedback.
Resources
- United States Pharmacopeial Convention, Inc. United States Pharmacopeia–National Formulary. USP Chapter <795>; USP Chapter <797>; USP Chapter <1163>; USP Chapter <61>; USP Chapter <62>; USP Chapter <1111>; USP Chapter <51>; USP Chapter <1207>. Rockville, MD: United States Pharmacopeial Convention, Inc.; Current Edition.
- U.S. Food and Drug Administration. Guidance Document: Container and Closure System Integrity Testing in Lieu of Sterility Testing as a Component of the Stability Protocol for Sterile Products Guidance for Industry. [FDA Website.] February 2008. Available at: www.fda.gov/regulatory-information/search-fda-guidance-documents/ contaIner-and-closure-system-integrity-testing-lieu-sterility-testing-component-stability-protocol. Accessed January 20, 2021.
- Vu N, Nguyen K, Kupiec TC. Quality-control analytical methods: The essentials of United States Pharmacopeia Chapter <51> Antimicrobial Effectiveness Testing and its application in pharmaceutical compounding. IJPC. 2014; 18(2): 123–130.
- Vu N, Lou JR, Kupiec TC. Quality control analytical methods: Microbial limit tests for nonsterile pharmaceuticals, Part 1. IJPC. 2014; 18(3): 213–221.
- Vu N, Lou JR, Kupiec TC. Quality control: Microbial limits tests for nonsterile pharmaceuticals, Part 2. IJPC. 2014; 18(4); 305–310.
James Zellner, ARL Bio Pharma Technical Sales and Microbiologist
USP <60>, Microbiological Examination of Nonsterile Products – Tests for Burkholderia cepacia Complex, describes a test procedure that evaluates the microbiological quality, specifically the presence of species of the genus Burkholderia, in non-sterile substances and preparations. This is important, because Burkholderia is a potentially dangerous pathogen, especially for drugs or raw materials that are intended for inhalation use, and aqueous preparations for oral, oromucosal, cutaneous, or nasal administration.
There are several clinical reasons why it is important to screen for this genus of microorganisms. First, they are Gram-negative, indicating their presence will produce endotoxins, potentially triggering a pyrogenic effect in a patient. Burkholderia is an opportunistic pathogen, commonly causing pneumonia in immunocompromised persons or those with existing lung diseases. Lung infection from Burkholderia is dangerous, and is characterized by a steep decline in function, potentially resulting in death. According to Lyczak (2002), death rates can be nearly 5x higher in Cystic Fibrosis (CF) patients infected with Burkholderia vs non-infected CF patients. This, combined with its ability to spread person to person via body fluids, generally results in strict isolation procedures if detected in the hospital setting. Burkholderia are naturally resistant to many antibiotics, including aminoglycosides and polymyxin B. Treatment generally requires a combination of several antibiotics. Combinations of ceftazidime, doxycycline, chloramphenicol, and others have been used as successful treatment options.
To test for Burkholderia, ARL Bio Pharma prepares enrichment media (Tryptic Soy Broth), along with a specialized Burkholderia cepacia Complex Selective Agar. This special agar inhibits non-Burkholderia organisms, and changes color to indicate the likely presence of the target microorganisms. Prior to use, the special agar media is checked to ensure it is inhibitory to Pseudomonas aeruginosa and Staphylococcus aureus and will indicate the presence of Burkholderia via a color change with 3 different species of Burkholderia. Once the quality of the media is verified, method suitability is performed to ensure the microbiologist is using a test procedure that will reveal Burkholderia if present in a test sample. After method suitability is demonstrated, evaluation of samples can begin. The test takes approximately 6-7 days and involves using the enrichment media first to encourage the growth of organisms in the sample, then the special agar to screen for the target organisms. Should potential Burkholderia species be detected via both growth and color change on the specialized media, microbial identification is carried out to confirm.
USP <60> is enforceable as a test for applicable products and raw materials. References to USP <60> can be found in many of the Not Yet Official sections set to be official August of 2021. This includes citations in Chapter Guides for Microbiology testing, Excipient testing, and Non-Complex APIs. USP <60> is already referenced in USP <1231> Water for Pharmaceutical Purposes as a potential contaminant in water systems. At some point, this chapter could be as commonly referenced in USP <61> and USP <62> for evaluation of the microbiological quality of raw materials and finished non-sterile products, especially those intended for administration orally, nasally, or for inhalation. Please contact ARL if you have questions about USP <60> testing and would like more information about submitting samples.
References:
- Lyczak, Jeffrey B et al. “Lung infections associated with cystic fibrosis.” Clinical microbiology reviews vol. 15,2 (2002): 194-222. doi:10.1128/cmr.15.2.194-222.2002
- Sousa, Sílvia A et al. “Burkholderia cepacia Complex: Emerging Multihost Pathogens Equipped with a Wide Range of Virulence Factors and Determinants.” International journal of microbiology vol. 2011 (2011): 607575. doi:10.1155/2011/607575
- USP <60> Microbiological Examination of Nonsterile Products Tests for Burkholderia Cepacia Complex
- USP <61> Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests
- USP <62> Microbiological Examination of Nonsterile Products: Tests for Specified Microorganisms
- USP <1111> Microbiological Examination of Nonsterile Products: Acceptance Criteria for Pharmaceutical Preparations and Substances for Pharmaceutical Use
- USP <1231> Water for Pharmaceutical Purposes
Evelyn Orona, Associate Microbiology Supervisor
Just as crucial as the sterile production of drug products is ensuring those same products are free of dangerous pyrogens – contaminants that induce febrile reactions in patients if introduced at high levels.
Relative to patient safety, the most concerning and common pyrogen is bacterial endotoxins. Pyrogens can be removed or destroyed by depyrogenation. A common method for depyrogenation is dry heat. Largely reproducible and easily controlled, dry heat depyrogenation is dependent on temperature and time to eliminate bacterial endotoxins. The depyrogenation process can be qualified and monitored with Endotoxin Challenge Vials (ECVs).
ECVs contain known concentrations of bacterial endotoxins and are available from multiple manufacturers. In addition to the physical measurements (temperature and holding time) of applicable equipment, ECVs are an excellent tool to use in confirming the effectiveness of a Dry Heat Depyrogenation cycle. Once a firm has conducted appropriate heat distribution studies in the oven being validated, ECVs can be placed in locations throughout an oven chamber, with specific focus on areas difficult to heat, known as “cold spots.” Following completion of the depyrogenation cycle, the oven’s ability to destroy endotoxins is measured by comparing the endotoxin concentration in test ECVs to the endotoxin concentration of untested (positive control) ECVs.
A log reduction in endotoxin concentration is calculated and used to determine whether the depyrogenation cycle is effective. A 3-log reduction is a commonly used benchmark to assess a deyprogenation process.
However, there may be circumstances which require a higher or lower log reduction. The appropriate endotoxin log reduction for a process should be determined based on several factors including:
- Native levels of endotoxin
- Efficiency of depyrogenation methods
- Patient safety
Manufacturers of ECVs generally list a 3-log reduction as the required specification. Firms can refer to USP <1228> Depyrogenation and <1228.5> Endotoxin Indicators for Depyrogenation for further guidance in determining a suitable log reduction specification for their processes.
At ARL Bio Pharma, when ECVs are received for testing, the samples are inspected and prepared per the ECV manufacturer’s instructions. Using Nexgen-MCS or the kinetic turbidimetric assay, Endotoxin testing is performed on each ECV.
Once results with passing quality control parameters are obtained, the Microbiologist calculates the log reduction between the test and control ECVs. All Certificate of Analyses generated include a test note indicating the log reduction achieved and whether it meets the ECVs’ manufacturer’s specification.
Performance of ECV testing should be scheduled periodically and is an important part of the depyrogenation process. For more information on Endotoxin Challenge Vial testing, contact ARL at 800-393-1595 or info@arlok.com.
Drug components differ in purity, specifications and quality. According to FDA draft guidance, it’s important for compounding pharmacies and outsourcing facilities to verify drug components purchased from suppliers.
Pharmacists verify drug components by:
- reviewing the supplier’s certificates of analysis (COA)s;
- establishing reliability of the supplier’s analyses through vendor qualification; and,
- testing drug components before use in compounding.
The FDA draft guidance for 503B outsourcing facilities states that “in lieu of testing each shipment of each ingredient, a COA can be accepted from the supplier and evaluated to determine whether a lot can be used, provided that the following conditions are met”:
- Pharmacists confirm the supplier’s test results, no less frequently than annually for active ingredients and every two years for other components, by performing full compendial testing using the applicable USP or NF monographs and/or supplier’s in-house methods.
- Pharmacists conduct at least one identity test to confirm the component purchased from the supplier.
Pharmacists are responsible for compounding preparations of acceptable identity (active ingredient and excipient), strength (concentration of the active ingredient), quality (safe, effective and acceptable to the patient), and purity (not contaminated with potentially harmful substances). The following drug component specifications are considered critical to ensure the quality of a finished drug product: identity, strength, purity, particle size, sterility, and endotoxin. Nonsterile components from suppliers must include microbial and endotoxin testing when used in the production of sterile drug products.
If a drug component is stored for a long period of time or has had exposure to air, heat or other conditions that might adversely affect the drug component, the ingredients need to be re-tested for identity, strength, quality, and purity.
If a drug component has an assigned expiration date, the pharmacist cannot extend the supplier’s expiration date by means of re-testing. If a drug component is labeled with a retest date, the material can be re-tested in compliance with specifications to ensure that it is still suitable for use.
Visit ARL’s website to learn more about the testing services available for drug components. To request a quote email: info@arlok.com.
Additional Resources:
- Allen, LA “The Art, Science, and Technology of Pharmaceutical Compounding”
- Pharmacy Compounding of Human Drug Products Under Section 503A of the Federal Food, Drug and Cosmetic Act
- Guidance for Industry Current Good Manufacturing Practice – Interim Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act
- USP <795>
- USP <797>
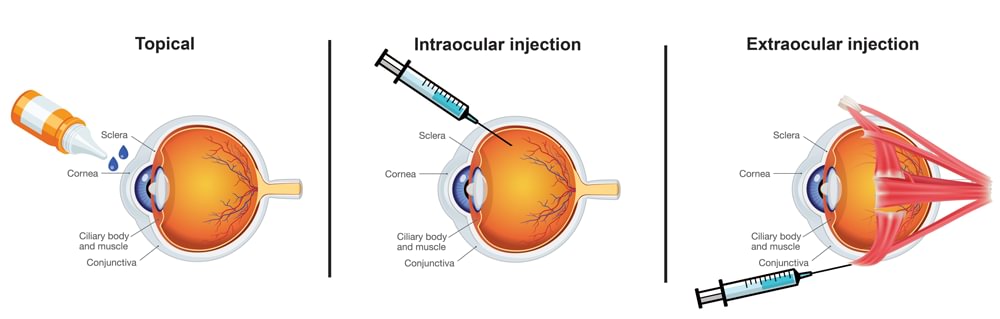
Ophthalmic medications must meet particulate matter test requirements. Particulate matter may come from foreign or product-related substances. Clinical exposure to unwanted particulate matter can cause a biological response in patients including blocking capillaries and arteries, introducing microorganisms and causing an infection, or causing an allergic reaction.
Ophthalmic products fall into three route of administration categories:
- Topical
- Intraocular Injections
- Extraocular Injections
USP <771> Ophthalmic Product Quality Tests states “all ophthalmic products should be inspected for package integrity, and to the extent possible, for the presence of observable foreign and particulate matter (visible particulates)”. USP <771> also establishes subvisible particulate matter limits based on two categories for product administration to the eye:
| <789> Particulate Matter in Ophthalmic Solutions | Intraocular administration which includes all ophthalmic products that cross (penetrate) boundary tissue, such as the cornea and sclera |
|---|---|
| <788> Particulate Matter in Injections | Extraocular administration which includes all other ocular components and spaces (Includes Topicals and Extraocular Injections) |
Ophthalmic preparations that are suspensions, emulsions, or gels are exempt from USP <789> requirements, as are medical devices.
Once the appropriate test method has been selected, procedures for the determination of particulate matter require that an ophthalmic solution must first be tested by the light obscuration procedure (Method 1). If it fails to meet the prescribed limits, it must pass the microscopic procedure (Method 2) with its own set of test limits. Where the ophthalmic solution cannot be tested by light obscuration, microscopic testing may be used exclusively if the light obscuration procedure has been demonstrated incapable of testing the solution or it produces invalid results.
Limits for Light Obscuration and Microscopy
For intraocular use – USP <789>:
| Method | ≥10 µm | ≥25 µm | ≥50µm |
|---|---|---|---|
| 1 | NMT 50 particles/mL | NMT 5 particles/mL | NMT 2 particles/mL |
| 1 | NMT 50 particles/mL | NMT 5 particles/mL | NMT 2 particles/mL |
For extraocular use – USP <788>:
| Method | Sample Volume | ≥10 µm | ≥25 µm |
|---|---|---|---|
| 1-a | >100mL | NMT 25 particles/mL | NMT 3particles/mL |
| 1-b | ≤100mL | NMT 6000 particles/container | NMT 600 particles/container |
| 2-a | >100mL | NMT 12 particles/mL | NMT 2particles/mL |
| 1-b | ≤100mL | NMT 3000 particles/container | NMT 300 particles/container |
ARL recommends clients specify the route of administration when submitting ophthalmic injections for particulate matter testing. If the route of administration is not included on the sample submission form, ARL will test the injection by the most stringent conditions – USP <789>.
For more information on particulate matter testing, contact ARL at 800-393-1595 or info@arlok.com.
Resources:
- USP <1> Injections and Implanted Drug Products (Parenterals) – Product Quality Tests
- USP <771> Ophthalmic Products Quality Tests
- USP <788> Particulate Matter in Injections
- USP <789> Particulate Matter in Ophthalmic Solutions
- USP <1788> Methods for the Determination of Particulate Matter in Injections and Ophthalmic Solutions
A cleaning validation demonstrates a pharmacy, hospital, or outsourcing facility’s cleaning procedure is effective and consistent in cleaning equipment and compounding surface areas. The Food and Drug Administration (FDA) expects firms to have written procedures (SOP’s) detailing the cleaning processes used for various pieces of equipment. According to the FDA, 21 CFR Part 211, “Equipment and utensils shall be cleaned, maintained, and, as appropriate for the nature of the drug, sanitized and/or sterilized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.” A cleaning validation is how one proves their cleaning SOPs are sufficient.
Cleaning is necessary to prevent three types of contaminations:
- Cross contamination with active ingredients
- Contamination with unintended drug components
- Microbiological contamination
Quality programs should have documented cleaning validations for cleaning procedures used in:
- ISO 5 and ISO 7 environments
- Non-sterile hazardous compounding areas
- Non-hazardous compounding areas
Cleaning validations should be conducted on the worst-case scenarios to determine if cleaning procedures are effective at removing residual drug or microorganisms from equipment and compounding surface areas. Without a documented cleaning validation, the pharmacy would not have scientific evidence that equipment and surface areas are cleaned sufficiently to ensure the safety of drug products.
ARL Bio Pharma offers cleaning validations so our clients can have this necessary documentation. For residual drug testing, the client submits their cleaning procedure to ARL along with direct surface sampling or rinse samples. Direct surface sampling is the recommended sampling method and allows the accessible areas to be evaluated for proper cleaning. Rinse sampling allows for larger surface areas and inaccessible systems or ones that cannot be routinely dissembled to be evaluated. For microorganism testing, a cleaning procedure is submitted to ARL along with coupons and cleaning agents.
Our laboratory develops the method for quantitation of active ingredients, unintended drug components, and/or microorganism detection. Next, ARL will simulate surface sampling to demonstrate that the our procedure can recover drug residue or microorganisms. The FDA does not set acceptance specifications or methods for determining whether cleaning processes are validated; however, it does recommend that residue and microorganism limits should be based on knowledge of materials and be practical, achievable, and verifiable.
Industry recommendations for acceptance specifications include:
- No more than 0.1% (1/1000th) of the normal therapeutic dose of any product will appear in the maximum daily dose of the following product, or
- No more than 10 ppm of any product will appear in another product, or
- No quantity of residue should be visible on the equipment after cleaning procedures are performed.
- Specifications and Limits according to USP <1072> Disinfectants and Antiseptics Guidance
Upon conclusion of the cleaning validation testing, ARL provides a final report. This report provides test results and any observations and conclusions regarding the effectiveness of the cleaning procedure. We also provide recommendations based on results found during the study.
For more information on cleaning validations, contact ARL at 800-393-1595 or info@arlok.com.
Resources for Cleaning Validations:
- FDA Guidance for Industry: Insanitary Conditions at Compounding Facilities
- USP <1072> Disinfectants and Antiseptics
- FDA Validation of Cleaning Processes (7/93)
- Current Good Manufacturing Practice – Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act
- USP <795> Pharmaceutical Compounding – NonSterile Preparations
- USP <797> Pharmaceutical Compounding – Sterile Preparations
- PIC/S Validation – Validation Master Plan Installation and Operational Qualification, Non-Sterile Process Validation, and Cleaning Validation
- Microbial Disinfectant Cleaning Challenge Studies
- ASTM – Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation
According to Center for Disease Control and Prevention (CDC), over 8 million US healthcare workers are exposed to hazardous drugs (HDs) every year. Drugs classified as hazardous include cancer therapy, antiviral drugs, hormones, and bioengineered drugs.
More than 12 billion doses of HDs are handled by US providers each year. Anyone handling HDs is at risk of exposure to acute and long-term effects including hair loss, cardiac toxicity, kidney damage, hearing loss, nausea, rashes, cancer, and infertility.
USP <800> provides standards for safe handling of hazardous drugs to minimize the risk of exposure to healthcare personnel, patients, and the environment. ARL Bio Pharma provides surface wipe sampling testing to measure the level of hazardous drug residue in areas where exposure can occur:
- Inside of C-PEC
- Pass through chambers
- Staging and work surfaces
- Floors and dispensing areas
- Outside HD buffer room or C-SCA
- Patient Administration
Surface Wipe Sampling evaluates your deactivating, decontaminating, disinfecting, and cleaning procedures. USP recommends sampling initially as a benchmark and at least every 6 months to verify containment of HDs.
The test is easy and inexpensive. Purchase a swab (TexWipe TX714K), swab area to be tested, collect sample, and submit sample for analysis.
Please do not submerge sample in a liquid, send swab only.
The cost is per swab – $200. One hazardous drug per swab.
A list of hazardous drugs (including hormones and cancer treatments) can be found in USP <800> and on the NIOSH Hazardous Drug List.
Available Hazardous Drugs for Testing
Contact ARL at info@arlok.com or 800-393-1595 for more information on Surface Wipe Sampling.