James Zellner, Microbiologist and Technical Sales Representative
What Growth Media is Appropriate for Specific Uses?
There are dozens of different types of microbial growth media available to Microbiologists, developed for a wide variety of purposes. The United States Pharmacopeia (USP) references many of these to demonstrate the quality of both compounded drug products and the control of compounding environments. The various types of media serve specific purposes based on the goal of the test. This might include recovery of the broadest range possible of organisms in a sterility test, screening for a specific organism if it would be particularly dangerous in a certain dosage form, or capturing anything in the compounding environment that might find its way into a finished product. Microbial growth media can be prepared in both liquid and solid forms, depending on the application. Tryptic Soy Agar (TSA) and Tryptic Soy Broth (TSB), for example, have the same nutrient profile and similar ingredients, but TSA is a solid and TSB is a liquid. For conducting passive air sampling, a solid TSA plate is easier to handle and leave out in the hood during compounding. Liquid media, like TSB, is generally better at growing organisms, and should be used where possible for recovery of microorganism in drug products. Liquid media is more conducive to growth, since nutrients, oxygen, and waste products move around more freely, and temperature is more uniform and constant in a fluid.
If the goal is to recover or grow the widest variety of organisms, a general growth media that provides a wide range of nutrients is the best choice. General growth medias mentioned in USP include TSB and TSA (aerobic), Fluid Thioglycolate Media (FTM, anaerobic), and Sabouraud Dextrose Broth and Agar (SDB/A, fungal). Tryptic Soy-based media provide a range of common nutrients for bacterial and fungal growth, and is referenced throughout the USP. If an anaerobic media is needed, FTM develops an oxygen gradient when left stationary, generating anaerobic conditions at the bottom and increasing oxygen levels at the top. Because of this property, FTM can grow both anaerobic (like Clostridium and Propionibacterium) and aerobic bacteria. SDB/A has a lower pH to encourage fungal and limit bacterial growth, making it an excellent general purpose fungal media.
General purpose medias are used to conduct USP <51>, USP <61>, USP <62> and USP <71> testing. In the case of USP <71> sterility testing, where any recovery is a potential Out-of-Specification result (simple pass/ fail criteria), general purpose media (TSB and FTM) may be the only type necessary. Other tests, like USP <62>, may start with TSB as an enrichment step, then move to more specialized media for the test to work properly.
Outside of testing product, TSA is referenced where environmental monitoring is discussed, since the goal is recovery of the broad range of organisms. TSA can also be incubated anaerobically, via Bio-Bag or specialized incubator, to recover anaerobic organisms.
In some cases, a general purpose media is not ideal for the test being conducted. A selective media might be used to exclude organisms if there is a specific species or group that is of interest. A USP example of this is Cetrimide Agar, used to screen for Pseudomonas aeruginosa in USP <62>. This agar contains a germicidal compound, Cetrimide, that P. aeruginosa and few other microorganisms can tolerate. If growth is observed on a Cetrimide Agar plate, it is almost certainly P. aeruginosa (though a microbial ID is recommended to confirm). Differential, sometimes referred to as indicative, media has some type of visual indicator added to quickly distinguish if a target organism is present. An example for this also comes from USP <62>, where MacConkey Agar is used to screen for Escherichia coli. MacConkey Agar contains lactose, which E. coli can ferment for energy, creating acid as a byproduct. A chemical additive in the media changes to a pink color as the pH lowers, providing the Microbiologist a visual cue that E. coli is likely present. Salmonella can also grow on MacConkey Agar, but cannot ferment lactose, so no color change will be observed. Finally, there are enriched growth media that contain additional nutrients, very specific combinations and amounts of nutrients, or additives to culture fastidious organisms (those with special growth requirements). USP <63> Mycoplasma refers to the use of enriched media for testing. Mycoplasmas are the smallest self-replicating bacterial organisms, and do not possess cell walls. They are of clinical concern because many are pathogenic, and can be a serious issue in laboratory and pharmacological cell cultures. The ability to screen for Mycoplasma is therefore extremely important. Their parasitic nature, very small genome, and lack of a cell wall mean they need very specific nutrient and growth conditions to be recovered in the lab. Hayflick Media, described in USP <63>, has additional nutrient sources added and antibiotics that act on organisms with cell walls (Penicillin) to meet the nutritional needs of Mycobacteria and exclude other bacterial organisms.
With the enormous range of microbiological media available, there is a “right tool” for many of the bacterial and fungal jobs in the lab. The USP provides a wealth of information and appropriate uses for the broths and agars encountered. An experienced Microbiologist, with the appropriate resources in equipment and media, is a powerful ally to ensure the highest quality and safest compounded drug products reach the patient.
For more information on growth media, contact ARL at info@arlok.com or 800-393-1595.
Andrew Taylor, ARL Bio Pharma Microbiology Supervisor
USP <795> Pharmaceutical Compounding – Non-sterile Preparations states compounders are responsible for minimizing patient harm resulting from multiple issues, including excessive microbial contamination in nonsterile drug products. The chapter directs compounders to USP <1163> for recommended quality control tests, including USP <61> Microbial Enumeration Tests and USP <62> Tests for Specified Microorganisms. The Food and Drug Administration (FDA) also refers to USP <1163> in 483 observations to compounding pharmacies for not performing microbiology testing of nonsterile products. Specifically referring to USP <61> to provide an estimate of viable aerobic organisms and USP <62> to demonstrate freedom from designated microbial species.
This article discusses the tests described in USP <61> and USP <62> that determine if non-sterile products meet quality requirements. These tests can also be used by sterile compounders for qualifying raw materials and performing in-process quality control testing.
USP <61> Microbial Enumeration Tests
USP <61> is often called a “Bioburden” or “Microbial Limits”. This test determines how many microorganisms are present in nonsterile drug products.
During a USP <61> test, the drug product is prepared and plated on two types of growth media, Soybean-Casein Digest Agar and Sabouraud Dextrose Agar. The plates are incubated at a defined temperature and duration. The number of colonies present on the plates are then counted and the results are calculated. To determine if the drug product passes the test, the number of colonies present in the drug product are compared to the acceptance criteria in USP <1111>.
USP <1111> provides acceptance limits for microorganisms present based on sample type (raw material or finished drug product) and route of administration.
In addition to USP <1111> Table 1, the significance of other microorganisms recovered should be evaluated in terms of:
- The use of the product: hazard varies according to the route of administration (eye, nose, respiratory tract)
- The nature of the product: Does the product support growth? Does it have adequate antimicrobial preservation?
- The method of application
- The intended recipient: risk may differ for neonates, infants, the debilitated
- Use of immunosuppressive agents, corticosteroids
- The presence of disease, wounds, organ damage
USP <62> Tests for Specified Microorganisms
USP <62> test results determine if objectionable microorganisms that could cause patient harm based on the route of administration are present in non-sterile drug substances or products. The microorganisms of concern listed in USP <62> include:
- Staphylococcus aureus
- Pseudomonas aeruginosa
- Salmonella
- Escherichia coli
- Bile-tolerant Gram-negative Bacteria
- Clostridia
- Candida albicans
A USP <62> test is initiated like USP <61> but uses microorganism specific growth media. At the conclusion of incubation, a result of “Pass” or “Fail” is generated. A passing result indicates the absence of the tested specified microorganism. USP <1111> provides absence of specified microorganism criteria based on sample type (raw material or finished drug product) and route of administration.
USP <61> and <62> are critical to ensure non-sterile drug products meet quality control criteria.
For more information on USP <61> and USP <62> testing, contact ARL at (800) 393-1595 or info@arlok.com.
Reference Documents:
- USP <61> Microbial Enumeration Tests
- USP <62> Tests for Specified Microorganisms
- USP <795> Pharmaceutical Compounding-Nonsterile Preparations
- USP <1111> Microbiological Examination of Nonsterile Products: Acceptance Criteria for Pharmaceutical Preparations and Substances for Pharmaceutical Use
- USP <1163> Quality Assurance in Pharmaceutical Compounding
FDA has identified the following list of drugs for the purpose of the temporary enforcement policies described in 503B guidance and 503A guidance during the COVID-19 public health emergency.
503B guidance establishes revised cGMP standards and allows outsourcing facilities to compound and distribute drugs needed to treat COVID-19 patients that hospitals cannot obtain from an FDA drug manufacturer due to drug shortage.
503A guidance allows pharmacies to compound drugs and distribute to pharmacies that are unable to obtain an FDA approved version of the drug or one compounded by a 503B outsourcing facility.
FDA recommends that State-licensed pharmacies consult with State authorities regarding local requirements.
Drugs eligible for compounding during the COVID-19 public health emergency
| Drugs | Currently in Shortage | Therapeutic Category |
| Cisatracurium besylate | Yes | Anesthesia |
| Dexmedetomidine hydrochloride | Yes, discontinuation reported | Anesthesia |
| Epinephrine | Yes | Cardiovascular; Pulmonary/Allergy |
| Etomidate | Yes, discontinuation reported | Analgesia, anesthesia |
| Fentanyl citrate | Yes | Analgesia, pediatric |
| Furosemide | Yes | Cardiovascular |
| Hydromorphone hydrochloride | Yes | Analgesia |
| Ketamine hydrochloride | Yes | Anesthesia |
| Lorazepam | Yes | Neurology |
| Midazolam hydrochloride | Yes | Anesthesia, neurology |
| Morphine Sulfate | Yes | Analgesia |
| Norepinephrine bitartrate | No | Cardiovascular |
| Rocuronium bromide | No, resolved | Anesthesia |
| Vancomycin hydrochloride | No | Antibiotic |
| Vecuronium bromide | Yes | Anesthesia |
| Dexamethasone sodium phosphate | Yes | Oncology |
Alliance for Pharmacy Compounding (APC) has established a bulletin board for 503B outsourcing facilities and 503A pharmacies to provide information to hospitals and hospital systems about which drugs they are currently compounding or able to compound for immediate distribution to hospitals in the states in which they are properly licensed. The information you provide will be made available to hospitals that are having difficulty sourcing COVID treatment drugs for their patients.
Andrew Taylor, Microbiology Lab Supervisor
Celsis rapid sterility testing is an alternative test method to USP Chapter <71>, which allows for shortened incubation times compared to the traditional sterility testing method. Where USP <71> requires between 14 and 18 days of incubation before a final test result, a Celsis rapid sterility test result can be generated after only 6 days of incubation. A reduction in test time of between 8 and 12 days offers a significant benefit when considering supply chain issues, product release schedules, and the timeliness of contamination investigations. The shortened incubation time is made possible by utilizing advanced technology designed to detect microbial growth much more quickly than visualized by the human eye.
ARL Bio Pharma uses the Celsis® rapid microbial detection system from Charles River, which is a growth-based sterility test that detects microbial contamination based on the presence of microbial Adenosine Triphosphate (ATP) in a sample. Because it is a growth-based test, it is more readily likened to the compendial USP <71> test which has been used for decades. Additionally, when comparing the Celsis® system to other rapid sterility technologies, this method allows for the processing of multiple sample types (solutions, oils, suspensions, etc.), and because it is non-destructive, species-level identification can be performed in the event of a non-sterile test result. Rapid sterility using the Celsis® system replaces the subjectivity of the traditional sterility test with an automated assay, measured by an instrument, while generating a numerical result which requires minimal analyst interpretation. One of the many reasons ARL chose the Celsis® system, is the test is initiated using the same methodology (i.e. closed membrane filtration) as a traditional sterility test.
Perhaps the most important facet of a rapid sterility test is the completion of a thorough method validation protocol. The Celsis® system for rapid sterility testing has been validated by the vendor (Charles River) and numerous labs around the world. Product validations using the Celsis® system have been approved by regulatory agencies worldwide, including the FDA. ARL Bio Pharma’s validation strategy is based on direction in USP <1223> Validation of Alternative Microbiological Methods, which is referenced as an appropriate guidance by USP <797>. ARL’s validation protocol includes all required parameters listed in USP <1223> including specificity, limit of detection, and repeatability. Additional parameters were assessed including linearity and the recovery of stressed/injured microorganisms, which is considered imperative as injured microorganisms are more representative of potential contaminants that may be present in preparations tested.
As with USP <71> sterility testing, method suitability is required to determine whether any inhibitory or interfering properties are present in a drug product that may prevent the accurate detection of ATP. Method suitability testing shows that the rapid sterility test method is valid for the specific drug product and reduces the possibility of a false negative or false positive result. Interfering properties can vary between drug products and components of a drug product formulation. Active ingredients, inactive ingredients, preservatives, and vehicles can all produce interfering properties. This highlights the importance that method suitability testing is performed when any change is made to a formulation. Just as it is for traditional sterility testing, rapid sterility method suitability is formulation specific. Rapid sterility testing, like the Celsis® system used by ARL Bio Pharma, offers numerous benefits compared to a traditional sterility test; most notably shortened incubation times and reduced subjectivity in results analysis. The wait for traditional sterility results may introduce unnecessary risk to the production process in the event of contamination, additional storage requirements, merchandise hold times, and delays to market. Charles River’s Celsis® rapid sterility test performed by ARL Bio Pharma delivers results in just 6 calendar days, allowing customers to quickly confirm the presence or absence of microbial contamination.
For more information on rapid sterility testing, contact us at info@arlok.com or 800-393-1595.
References
– USP <797> Pharmaceutical Compounding—Sterile Preparations
– USP <1223> Validation of Alternative Microbiological Methods
Kathy Heatherly, MSFS, Technical Sales
As a compounder, protecting yourself and your staff from exposure to hazardous drugs should be important both from a safety and monetary perspective. USP <800> applies to sterile and non-sterile compounding pharmacies and anywhere that hazardous drugs are received, stored, or administered. It is also designed to prevent compounders from incurring fines from OSHA (Occupational Safety and Health Administration) for not providing a safe workspace and not complying with the information in USP <800> or on the NIOSH list. Compliance with regulations includes having: a negative pressure lab to compound hazardous drugs, the appropriate design, layout, or equipment in the pharmacy to compound these materials, and appropriate controls for handling finished dosage forms. If a facility is compounding or handling hazardous drugs in a state not enforcing USP <800> but is shipping to a state that is, they must comply. Even if a state board of pharmacy has chosen not to adopt USP <800>, OSHA is still the regulating agency and can inspect a facility that handles or compounds hazardous drugs and levy a fine if they do not comply.
Wipe sampling is best practice. Environmental wipe sampling for hazardous drug surface residue should be performed to verify containment. Contamination in any amount indicates a lack of containment. Although described in as a “SHOULD” versus a “MUST,” some type of environmental wipe sampling is strongly recommended. A facility cannot assess its success in controlling environmental contamination if it cannot measure the extent of such contamination. The testing that ARL offers is easy and inexpensive to do. Purchase a swab (TexWipe TX714K), swab area to be tested, collect sample, and submit sample for analysis. The cost is per swab – $200. One hazardous drug per swab. A list of hazardous drugs (including hormones and cancer treatments) can be found in USP <800> and on the NIOSH Hazardous Drug List.
Avoid OSHA fines and reduce potential employee claims for an unsafe workplace by testing with ARL.
For more information on USP <800> Surface Wipe Sampling Test, visit www.arlok.com/USP800
Which one is the most reliable for sterility testing?
According to USP <71> Sterility Tests, membrane filtration is the sterility test method of choice for filterable pharmaceutical products. Membrane filtration refers to either an open membrane filtration (OMF) or closed membrane filtration system. Not all sterility test systems are the same. It is important pharmacists know the differences between the two systems to reduce the risk of false positive results, costly investigations, and batch loss.
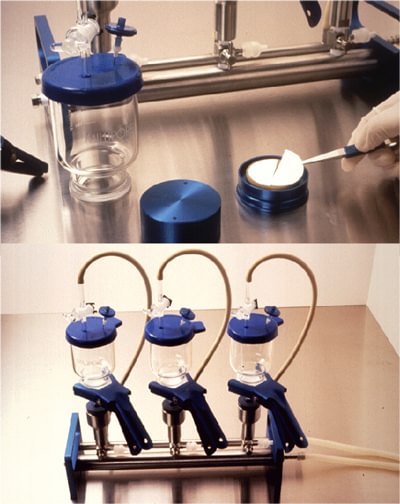
Open Membrane Filtration (OMF)
The OMF testing process passes the drug product through an open funnel containing a filter. Once appropriate filter rinsing is complete, the filter is removed, cut in half, and placed individually into each media type used for sterility testing; tryptic soy broth (TSB) and fluid thioglycollate medium (FTG or FTM). Incubation occurs at the appropriate temperatures and duration. The required handling of the filters greatly increases the risk of contaminating the sterility test sample during the sterility test procedure
Risks of using OMF:
- Sterile drug and sterility test filter are exposed to test environment increasing false positive results
- Drug product sample may pass around the removable filter and not through it causing a false negative
- Test operator has direct contact with filter membrane and must carefully cut and manipulate filter using scissors and tweezers to avoid contamination
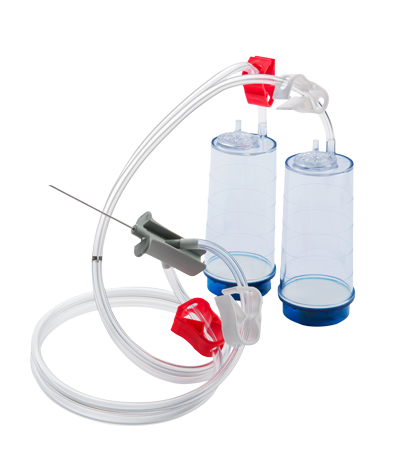
Closed Membrane Filtration
Sterility testing via closed membrane filtration is the most reliable method. It utilizes a dual canister system, where each canister contains its own 0.45 micron filter. Both canisters are attached to a single needle via sterile tubing. The test is carried out by creating a vacuum in the canisters, which allows the microbiologist to withdraw sample directly from the drug product being testing using the attached needle. The sample is then automatically split between both canisters and passes through both filters. Following the appropriate filter rinse, each media type (TSB & FTG) are individually added into the separate canisters. The tubing is sealed, and each canister is incubated at the appropriate temperatures and duration. Closed membrane filtration uses a secure system to provide high-quality sterility test results.
Benefits of using closed membrane filtration:
- The sealed drug product is never opened or pooled before filtration
- The filter itself is never opened or exposed to the test environment decreasing the risk of false positive results
- The filter is sealed in the canister reducing the risk of false negative results
- The test operator cannot touch or manipulate the filter membrane decreasing the risk of contamination
- All materials used are pre-sterilized and assembled, reducing possibility of test fault or contamination of testing materials by the laboratory
- The needle (attached to the filter canister unit) allows analysts to withdraw product for testing in the same way health professionals withdraw the product with a syringe
Sterility testing is an integral part of routine quality control and product release. It is important pharmacists use closed membrane filtration, the most reliable method, to reduce the risk of false positive results, prevent costly investigations and batch loss, and protect their patients from false negatives. ARL Bio Pharma only utilizes pre-sterilized, ready-to-use, closed membrane filtration canisters to ensure the highest quality sterility test for filterable products.
For more information about sterility testing, contact ARL at 800-393-1595 or info@arlok.com
Kathy Heatherly, MSFS, ARL Technical Sales
A bulk substance, also known as an active pharmaceutical ingredient (API), is often the starting point of a compounded preparation. Prior to use, compounding pharmacies and outsourcing facilities should confirm the quality of an active pharmaceutical ingredient to ensure consistency of the material as received from the supplier and suitability for use in a compounded preparation. At a minimum, identity testing is recommended for each lot, but more rigorous testing should be evaluated.
The United States Pharmacopeia, Food and Drug Administration, and International Council on Harmonization have published guidelines for testing APIs. Per USP General Notices, 4.10.10. Applicability of Test Procedures, “A single monograph may include more than one test, procedure, and/or acceptance criterion for the same attribute. Unless otherwise specified in the monograph, all tests are requirements. In some cases, monograph instructions allow the selection of tests that reflect attributes of different manufacturers’ articles, such as different polymorphic forms, impurities, hydrates, and dissolution.”
Per the cGMP Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act Guidance for Industry, published Dec 2018, “Components that are not approved finished drug products must be tested to verify identity and evaluated for conformity with appropriate specifications.” An objective of the ICH Good Manufacturing Practice for Active Pharmaceutical Ingredients Q7 is to provide guidance for assuring API’s meet the quality and purity requirements that the manufacturer says they possess. Sections 7.30 and 7.32 state, “For incoming production materials, identity tests and related methods should be used as described in the relevant sections of a pharmacopoeia monograph…The visual examination of a label or the material is not considered sufficient except where justified for processing aids, hazardous or highly toxic materials, other special materials…”
Potency, Sterility, and Endotoxin testing performed on the finished product are not enough to detect changes that would indicate suitability of the API for use in compounding. However, a test for identity only, may not provide enough information to ensure the API is fit for use in a finished product.
Additional compendial test methods that should be evaluated include organic and inorganic impurities, enantiomeric purity, residual solvents, volatile matter, assay, and microbial enumeration. Organic Impurities, both known and unknown can affect the quality, safety, and efficacy of drugs and should be controlled to avoid adverse effects in the finished product. Elemental or inorganic impurities have very low permissible exposure limits in a finished product. Verifying these impurities have been controlled is critical in ensuring that a product is suitable. Control of enantiomers and/or isomers is also critical; as changes to their form may result in no physical change in appearance or the compounding process, but could result in a marked difference in pharmacology, toxicology, pharmacokinetics, metabolism, etc. of the finished product.
With varying exposure limits depending on the route of administration and no therapeutic benefit to the finished product, the manufacturer’s results for residual solvents should be tested to ensure the material confirms to safety-based limits, ingredient and product specifications, and good manufacturing practices. Volatile matter and assay value of the API to be weighed and used in compounding must be known. Many of the USP monographs provide specifications for calculating the assay value that should be used when compounding a finished preparation. If the substance is a hydrate, its anhydrous equivalent weight may need to be calculated. On the other hand, if there is adsorbed moisture present, the weight of anhydrous drug substance may need to be calculated. In addition to testing when the material is sourced, if there have been multiple entries into the container over the life of the material, it is also recommended to test the moisture content again; as additional moisture can affect potency of the finished product. Testing of bioburden can confirm a controlled manufacturing process for API to be used in non-sterile preparations as well as verify the sterility assurance process for use of the API in a sterile finished product.
Identifying the correct material has been sourced, verifying that the specifications are met, and the material is safe for use in finished preparations ensures the quality of a product that is used in compounding the finished preparation.
For more information, contact ARL at 800-393-1595 or info@arlok.com.
Andrew Taylor, ARL Bio Pharma Microbiology Lab Supervisor
Pyrogens are a group of fever causing substances which can be found in compounded sterile preparations (CSPs) if appropriate actions are not taken to reduce or remove them. One of the most widely known groups of pyrogens are bacterial endotoxins.
Bacterial endotoxins are:
- Remnants of bacterial cells that can cause fever, diarrhea, or septic shock in patients
- Not removed during filter sterilization
- Not removed during steam sterilization
- Not detected by a sterility test
USP <797> requires a Bacterial Endotoxin Test (BET) for each preparation of:
- Category 2 and Category 3 CSPs compounded from one or more nonsterile component(s)
- Multiple-dose CSPs
USP <797> also requires a description of the depyrogenation process employed, including the temperature, pressure (if applicable), duration, permissible load conditions for each cycle; and the use of endotoxin challenge vials (ECVs) must be included in the facility’s SOPs. The chapter also states that if a CSP is dispensed or administered before Endotoxin testing results are known, a facility must have procedures in place to Immediately notify the prescriber of a test failure with the potential to cause patient harm.
The Food and Drug Administration (FDA) requires a BET for all 503B outsourcing facility drug products reported to be non-pyrogenic.
Even if it is not mentioned specifically in the regulatory documents, it is important to check for the presence of endotoxins in raw materials, at various points in the compounding process, and in finished products before administering a drug to a patient.
The limit for the maximum amount of endotoxin allowed to be present in a CSP can be found in the applicable USP monograph, or calculated using the route of administration, patient weight, and maximum bolus dose. Once the limit has been established, the primary test method used to determine the amount of bacterial endotoxins in a CSP is described in USP <85> Bacterial Endotoxins Test.
ARL Bio Pharma’s Bacterial Endotoxin Testing Process
ARL Bio Pharma follows the procedures described in USP <85>. The test is performed using depyrogenated glassware and supplies and the product to be tested is prepared using reagents specifically designed for BET. Each sample is tested in duplicate, using a standard curve, negative control, and positive control. At the conclusion of the test, the amount of endotoxin is calculated for each sample analyzed. If the endotoxin concentration is found to be below the limit, and all quality control parameters of the test are met controls were acceptable, the sample complies with USP <85> requirements.
Dr. Qiang Liu, Research and Development Laboratory Supervisor
A beyond use date (BUD) is the date after which a compounded preparation shall not be used. The BUD is determined from the date the preparation is compounded. This date should be based on drug-specific, scientifically valid studies when possible. Things to consider when assigning BUD include:
- The drug and its degradation mechanism
- The dosage form and its components
- The potential for microbial proliferation
- The container in which the preparation is packaged
- The expected storage conditions
- The intended duration of therapy
USP <797> describes four methods for assigning a beyond use date:
- Product labeling
- Commercial product manufacturer consultation
- Appropriate literature
- Direct testing
Beyond use dating must be carefully interpreted with respect to the actual compounded formulation and conditions for storage and use. Predictions based on literature are considered theoretical beyond-use dates as the published data introduces varying degrees of assumptions with a likelihood of error or inaccuracy. Pharmacists using literature to assign a beyond use date must look for the exact drug formula, storage conditions, and container/closure to reduce the likelihood of errors. State and federal regulations also require pharmacists to have written justification for a beyond use date assignment.
The only truly valid beyond use date is obtained through product-specific studies supported by scientific data. These direct testing studies use stability indicating methods (SIM) to ensure therapeutic effectiveness of compounded drug products.
A SIM is a reliable, meaningful, and specific analytical procedure that accurately and precisely measures active pharmaceutical ingredients (API) by separating the API from its degradation products and excipients. A SIM must be validated for the exact formulation being tested. High performance liquid chromatograph (HPLC) is one of the most commonly used techniques for examining the chemical stability of compounded product, but not all HPLC tests are stability indicating. A forced degradation study must be performed on the compounded drug product and not inferred from testing on only the API.
Testing the concentration of the drug is just one component of a stability study. Testing should include evaluation of the physical, chemical, and microbiological properties of the product. Common tests in a stability study include:
- Assay (Stability Indicating Method)
- Sterility – USP <71> (sterile preparations)
- Endotoxin – USP <85> (sterile preparations)
- pH – USP <791>
- Visual Inspection (Appearance)
- Particulate Matter – USP <788> / <789> (all sterile solutions for IV injection or ophthalmic)
- Preservative Effectiveness – USP <51> (preparations containing an antimicrobial preservative)
- Preservative Quantification – (preparations containing an antimicrobial preservative)
- Microbial Limits – USP <61> (nonsterile preparations)
- Absence of Specified Organisms – USP <62> (nonsterile preparations)
Additional tests for biological products BUD include:
- Protein content – USP <1057>
- Potency or activity – such ELISA assay
- Product related impurity including protein aggregates, size and charge variants
Please contact ARL (800) 393-1595 or info@arlok.com with questions.
Qiang Liu, PhD, Research and Development Supervisor
Container Closure Integrity Testing (CCIT)
The container closure system for a drug product provides critical protection for stability and sterility. Contaminants (such as microorganisms and reactive substances) could potentially cross through defective container closures putting the drug product and patients at risk. USP chapter <1207> defines the concept of container closure integrity as “encompassing the absence of all package leaks that risk product quality”. FDA also introduced container and closure system integrity testing in lieu of sterility testing as a component of a stability protocol for sterile products.
Among a variety of CCIT tests, dye ingress testing is one of most commonly used methodologies. During the test, the container closure test articles are put into an empty vacuum chamber to introduce negative pressure to the articles. This vacuum removes air inside the container/closure system. The vacuum is then released and methylene blue hydrate solution enters the chamber until it reaches atmospheric pressure. The articles are removed from the chamber, rinsed, patted dry, and examined for the presence of methylene blue through visual inspection or spectrometry. To pass the test, there must be no detectable methylene blue inside any of the tested container closure systems. ARL provides CCIT services for a variety of container types and compounds including colored compounds such as Methylcobalamin, Cyanocobalamin, and Mitomycin.
The sensitivity and applicability of CCIT methodologies varies based on container type, size, fill volume, and compounded drug product. ARL recommends and provides CCIT method validation services for clients’ specific container closure system.
Please contact ARL for more information on CCIT 800-393-1595 or info@arlok.com.