ARL’s sterility testing cleanroom provides reliable results to verify your people, processes, and drug products are free of contamination. This pharmaceutical grade facility has cutting edge technology that controls the sample test environment and improves the reliability of your sterility results. Our cleanroom provides you faster results in a controlled environment you can trust.
Call 800-393-1595 today with questions or to schedule a tour of our facilities.
Kerri Hirst, Senior Microbiologist
Sterility testing by USP <71> is a test used in tandem with other sterility assurance procedures and tests to ensure that a product is free of microbial contaminants and safe for patients to use. For a sterility test to perform appropriately, a method suitability validation must be completed on each specific formulation to determine the appropriate test method.
Sterility method suitability testing is performed to determine whether any inhibitory or antimicrobial properties in a drug product will prevent the sterility test from detecting the presence of viable microorganisms. It shows that the sterility test method is valid for the specific drug product and reduces the possibility of a sterile result on a product that is not sterile. Inhibitory properties can vary between drug products and components of a drug product formulation. Active ingredients, inactive ingredients, preservatives and vehicles can all produce inhibitory properties. This highlights the importance that method suitability testing be performed when any change is made to a formulation. Method suitability is formulation specific!
A sterility test method is selected based on the known inhibitory nature of a formulation based on a detailed evaluation of the formulation sheet, including applicable sub-formulations. For a closed membrane filtration method, a volume which is greater than or equal to the volume to be used during the sterility test is passed through two closed system filtration devices. Viable microorganisms will be retained on the filters while the drug product is transferred to a waste container.
The filters are rinsed using fluid A, D or K to remove any lingering drug product from the filters and containers. Fluid thioglycollate medium (FTG) and tryptic soy broth (TSB) are filled independently into each container. Membrane filtration is the preferred method for the sterility test by USP <71> because it separates the product from the potential microbial contaminants which reduces or eliminates the inhibitory properties of the drug. Another test method, direct inoculation, where the product is diluted directly into FTG and TSB to overcome the inhibition of the product, is used when the product to be tested is unfilterable. Certain oils used for vehicles or products that are suspension mixtures can make filtration impossible. The chosen test method is performed using the drug product in 3 TSB and 3 FTG containers, where six test organisms are inoculated into the applicable media, incubated for up to 5 days, and observed for growth.
The sterility method suitability test organisms are a range of different types of organisms that require different types of growth conditions and are representative of common organisms that may be found in the compounding environment.
| Organism | Type | Properties | Oxygen? | Media |
|---|---|---|---|---|
| Staphylococcus aureus | Bacteria | Gram-positive cocci (Spherical) | Aerobic | FTG |
| Bacillus subtilis | Bacteria | Gram-positive Rod, Spore forming | Aerobic | TSB |
| Pseudomonas aeruginosa | Bacteria | Gram-negative Rod | Aerobic | FTG |
| Clostridium sporogenes | Bacteria | Gram-positive Rod, Spore forming | Anaerobic | FTG |
| Candida albicans | Fungi | Budding yeast | Aerobic | TSB |
| Aspergillus brasiliensis | Fungi | Spore forming filamentous mold | Aerobic | TSB |
If all of the organisms grow in the same manner as the positive control and no growth is observed in the negative control, the method suitability has passed and the method may be used for the sterility test. If all the test organisms do not grow, the test must be repeated using a new method that utilizes additional neutralizing practices. Increasing the ability of a test method to neutralize the antimicrobial properties can be done in several ways. For overcoming the inhibition of more antimicrobial products such as antibiotics using closed membrane filtration, a pre-filtration dilution into fluid A can be added. Then, after the diluted sample is passed through the filter, a secondary rinse can be performed. For direct inoculation methods, increasing the dilution or adding USP-approved neutralizers to the media to overcome the inhibitory properties of the product can be done. The appropriate sterility test method can be determined by trying one or more attempts using increasingly more conservative test methods until all six organisms are recovered.
The method determined to be suitable for a specific formulation directly impacts the confidence in all future sterility tests for that product. The formulation must stay the same for the method to be valid for the sterility test on future lots. Changes to any of the components of the formulation or sub-formulation(s) can impact the validity of the sterility test, and method suitability must be repeated. The sterility test is a critical part of the sterility assurance level required by compounders, but it is not the only indicator of sterility assurance. It is up to the compounding facility to develop a sterility assurance plan which should include the sterility test and it is up to ARL Bio Pharma to provide the best sterility test service to meet the need for our clients.
For more information, contact ARL at 800-393-1595 or info@arlok.com.
James Zellner, ARL Bio Pharma Technical Sales
How do you know your staff is doing a thorough job disinfecting surfaces and equipment at your facility? Do you have proof that your cleaning procedure is effective in removing potential contaminants from surface areas?
An important part of a complete quality program is generating data that the standard operating procedures (SOPs) in place are achieving their intended goals. Specific to cleaning SOPs, pharmacies must demonstrate that cleaning agents, combined with instructions and procedures for their use, remove microbial contamination from surface areas where sterile drug products are compounded. USP <1072> Disinfectants and Antiseptics states explicitly in its first line,” A sound cleaning and sanitization program is needed for controlled environments used in the manufacture of Pharmacopeial articles to prevent the microbial contamination of these articles”. This USP directive is met by performing Microbial Disinfectant Challenge studies.
There are two parts to consider when designing a disinfectant challenge study to generate data that demonstrates the efficacy of a facility’s cleaning SOP and choice of agents for removing microbial contamination. Use-Dilution testing ensures that agents at their in-use concentrations, whether premixed or mixed on-site, can decontaminate a surface in the contact time specified. Once the efficacy of the cleaners is established, Surface Challenge testing determines the effectiveness of procedures to remove contamination from appropriate surfaces. The agent’s individual efficacy is demonstrated with Use-Dilution testing, followed by combined usage by SOP instruction in the Surface Challenge test.
Cleaning studies should have sound and realistic acceptance criteria based on the cleaner’s method of action, surface cleaned, organism(s), and contact time. A well planned microbial disinfectant cleaning study provides a facility the information to confidently continue using their selected methods of decontamination, or data that indicates a modification to SOPs is necessary to improve the state of control.
While the specifics of a microbial cleaning challenge study may vary, a general framework is recommended. Three lists are the driving factors behind the study design. The examples below are not exhaustive, but provide the cleaners, surfaces, and organisms to use. First, make a list of the cleaners used in your facility. The cleaning agents should complement each other’s method of action and fall into several groups:
- Hypochlorite
- Phenolics
- Peroxides
- Alcohols
- Sporicides
Generally, a combination of disinfectants with germicidal and sporicidal properties are used in sequence with a final sterile alcohol step during cleaning.
Once the cleaners and procedures for their use are specified, list the surfaces to clean. Commonly cleaned surfaces in the compounding pharmacy are:
- Stainless Steel (the grade should be specified)
- Glass
- Plexiglass
- Aluminum
- Clear lighting panels
- Epoxied floor
- Ceiling tiles
- Wall surfaces
- Flooring surfaces
Facilities should acquire coupons (small samples) of each of the surfaces from the manufacturer or another source for submission to a laboratory for surface challenge testing. The lab uses the facility’s cleaning SOPs and mimics the surfaces cleaned at the compounding facility.
Next, select microorganisms to verify the cleaners and procedures. In USP <1072>, Table 5 provides a guide for organism selection. Like many of the microbiological chapters in USP, the organisms chosen should be representative of a class of potential contaminants. Suggested bacteria and fungi that serve as challenge organisms are:
- Staphylococcus aureus (Gram-positive cocci)
- Pseudomonas aeruginosa (Gram-negative rod)
- Escherichia coli (Gram-negative rod)
- Bacillus subtilis (Spore-forming bacteria)
- Candida albicans (Yeast)
- Aspergillus brasiliensis (Mold)
A facility may also use bacterial and fungal organisms recovered during environmental monitoring as challenge organisms. This is important if an environmental recovery is of a class not covered by the traditional representative organisms mentioned above.
Finally, acceptance criteria needs to be clearly stated. USP <1072> gives an example of a 2-log reduction in bacterial spores and a 3-log reduction in vegetative bacteria on the coupons used in the Surface Challenge test. This example expectation can be further expanded based on the cleaning agent’s efficacy and the organisms tested. Ultimately, the facility and lab should work together to establish acceptance criteria for the cleaning agents and methods. If changes are made to the SOP, as part of a continuous effort to maintain the optimal compounding environment, an additional study should be performed to ensure that the new procedure meets the criteria of the tests.
Microbial cleaning studies are essential to validating a facility’s cleaning SOP. This is an important component to ensure quality drug products reach your patients. It demonstrates that the disinfecting steps and agents a compounder is taking to maintain controlled areas are adequate to produce a sterile product.
For more information, contact ARL at 800-393-1595 or info@arlok.com.
Cindy Pickens, ARL Bio Pharma Laboratory Supervisor
Particulate Matter testing is performed on all parenteral solutions to determine the cleanliness and stability of the solution; and, is listed in USP <797> as a necessary component for quality assurance of compounded sterile preparations under the responsibilities of compounding personnel. It is also specifically outlined as a required test in several individual monographs such as Morphine Sulfate Compounded Injection.
USP Chapter <788> specifically deals with particulate matter in parenteral solutions. The analysis can be carried out in either two methods and are further broken down into categories, ‘a’ and ‘b’, based on the volume of the sample. Method 1 is an instrumental method that utilizes light obscuration on a prescribed volume of solution and is the preferred method of analysis. Method 2 is a manual microscopic method where the sample is manually filtered through a membrane and physically counted utilizing a microscope. Method 2 is used only when Method 1 cannot be performed. Examples of samples that must be analyzed by Method 2 are those that are oil, highly surfactant, or darkly colored. Each method is broken down into category ‘a’ and ‘b’. Samples in containers >100mL are analyzed by category ‘a’ which provides specifications as “particles/mL”. Samples in containers ≤100mL are analyzed by category ‘b’ which provides specification as “particles/container.” Specifications are:
| Method | Sample Volume | ≥10 µm | ≥25 µm |
|---|---|---|---|
| 1-a | >100 mL | NMT* 25 particles/mL | NMT 3 particles/mL |
| 1-b | ≤100 mL | NMT 6000 particles/container | NMT 6000 particles/container |
| 2-a | >100 mL | NMT 12 particles/mL | NMT 2 particles/mL |
| 2-b | ≤100 mL | NMT 3000 particles/container | NMT 300 particles/container |
*NMT = “Not More Than”
As seen from the specifications, the volume of the sample dictates the format of the results (‘per mL’ or ‘per container’). USP dictates that a minimum of 25mL of sample is required to run a particulate matter test. As long as the sample container has a minimum of 25mL, the sample can be run without further consideration. However, if the container volume is less than 25mL, USP requires that 10 containers must be combined for analysis. If the combined volume of 10 containers is less than 25mL, the sample can be diluted with particle free water so the analysis can be conducted. A dilution factor is applied to allow for an accurate particles/container count.
USP Chapter <789> deals specifically with ophthalmic solutions. In principle, the test is performed in the same manner as <788> Method 1; however there is only one set of limits for ophthalmic solutions regardless of the volume of the sample unless the monograph for that solution dictates otherwise. Additionally, <789> calls for additional testing via Method 2 if it first fails by Method 1.
| Method | ≥10 µm | ≥25 µm | ≥50 µm |
|---|---|---|---|
| 1 | NMT 50 particles/mL | NMT 5 particles/mL | NMT 2 particles/mL |
| 2 | NMT 50 particles/mL | NMT 5 particles/mL | NMT 2 particles/mL |
USP Chapter <787> can be used as an alternative for <788> for therapeutic protein injections. This method allows for a lower volume of material to be utilized for testing. The test is performed by Light Obscuration (Method 1) with special consideration given for the sample type, additional system suitability verification steps and blank readings. While some of these samples can be run by method 2, the results are not considered interchangeable as with <788> and <789> due to visual interference of the protein particles and their physical characteristics. The specifications for <787> are equivalent to <788>.
| Method-category | Sample Volume | ≥10 µm | ≥25 µm |
|---|---|---|---|
| 1-a | >100 mL | NMT 25 particles/mL | NMT 3 particles/mL |
| 1-b | ≤100 mL | NMT 6000 particles/container | NMT 6000 particles/container |
It is important for pharmacists to conduct particulate matter testing on parenteral solutions, as particulate matter in large numbers can cause harm to patients. Testing per USP methods assures compounding personnel that their products meet the requirements for sub-visible particles.
Please contact ARL for more information 800-393-1595 or info@arlok.com.
Jessica Munson, M.S., ARL Bio Pharma Analytical Supervisor
What is a Master Formulation Record?
A Master Formulation Record is used to document the specific information for each individual batch and is an important component of regulatory compliance and effective process control. This detailed record of procedures describes how the drug product is to be prepared. According to USP <795> and <797>, a Master Formulation Record must be created for:
- each unique formulation of a compounded nonsterile preparation (CSNP)
- compounded sterile preparations (CSP) for more than 1 patient
- CSP from nonsterile ingredient(s)
Visit USP <795> and <797> for more information on a Master Formulation Record and its required contents.
Master Formulation Records and Potency Testing
It’s important for a pharmacy to receive potency results in the time requested. Submitting a Master Formulation Record with every sample helps improve on-time delivery to the pharmacy and reduces potential out-of-specification (OOS) investigations. This allows for any discrepancies to be caught during accessioning of the sample, analysis of the sample, and/or reporting of the results. Thus, reducing the need for additional client communication to answer questions that might arise. Some questions that can quickly be answered using the Master Formulation Record are:
- What form of Active Pharmaceutical Ingredient (API) is to be reported (i.e. Fentanyl vs. Fentanyl Citrate)?
- Was the formulation compounded using raw material, a triturate, or a commercial product?
- Was water content/LOD accounted for during compounding?
- Was the product formulated on a per weight basis or a per volume basis?
Master Formulation Records are essential to providing the laboratory necessary information on how the drug product is prepared. It gives a thorough view of your pharmacy’s processes and aids our laboratory in providing you high-quality results.
For more information about submitting Master Formulation Records, contact ARL at 800-393-1595 or info@arlok.com.
Abstract:
Potency tests, known as quantitative tests, are designed to determine how much of the active drug is in the sample. Stability tests are used to determine a beyond-use date for a preparation. Employing the proper method to determine potency or stability is key to understanding the difference between potency testing and stability testing. Methods of determining potency may or may not be stability indicating, but stability can be determined only by a stability-indicating method. A stability-indicating method can determine both potency and stability. Quality assurance programs are essential to establishing standards for compounded preparations. It is important that compounding pharmacists understand the differences between potency and stability tests and that these tests are made an integral part of the quality assurance program.
____________________________________________________________________________________
Oftentimes the question is asked “What is the difference between potency and stability?” This seems like a rather simple question and in some respects it is. To answer this question, however, the differences in the methods used to analyze the potency and stability of a compound must be understood. The most common mistake in determining stability is failure to use an analytical method that has been demonstrated to be stability indicating.1 It is not a surprise, therefore, that the most important aspect of determining potency and stability is the methods employed in the process. Simply put, a stability-indicating method must be used to determine stability. A stability-indicating method also can determine potency, but not all potency tests can determine stability.
The purpose of this article is to explain the difference between potency and stability, why they are important, and how they are determined. The method used to determine the concentration of the active ingredient, or analyte, is the most critical step in the process.
Quality Assurance in Compounding
United States Pharmacopeia (USP) Chapter <1075> Good Compounding Practices defines compounding as “the preparation, mixing, and assembling, packaging, and labeling of a drug or device in accordance with a licensed practitioner’s prescription of medication or under an initiative based on the practitioner/patient/pharmacist/compounder relationship in the course of professional practice.”2 The art of pharmaceutical compounding has long been a fundamental element in the profession of pharmacy.3 In an effort to ensure that each preparation is made appropriately and safely, a set of standards has been developed by the United States Pharmacopeial Convention, Inc. (USP). One such standard, included in Chapter <1075>, is the requirement that a compounded preparation be assigned a beyond-use date.2 The beyond-use date must be based on published data, appropriate testing (i.e., stability-indicating method), or USP–National Formulary standards.2 It is the compounding pharmacist’s responsibility to follow the USP guidelines when preparing compounded medications. Chapter <1163> Quality Assurance in Pharmaceutical Compounding defines a quality assurance program as “a system of steps and actions taken to ensure the maintenance of proper standards in compounded preparations.”4 A quality assurance program is essential to ensuring that the USP guidelines are met and that each compounded preparation is safe.
When compounding a new preparation that has not been tested appropriately, it is important that potency and stability studies are performed to determine concentration and a beyond-use date, respectively. A medication that is very potent (e.g., fentanyl) or has a narrow therapeutic index (e.g., levothyroxine) may produce a magnified (or greatly reduced) effect if its concentration is altered even slightly. Nitroglycerin, an antianginal medication that patients rely on to avoid chest pain, has a questionable stability profile. In the event that the beyond-use date is not accurate, and the patient does not experience relief because the medication is inactive, the consequences could be severe, including hospitalization and even death. Although these drugs may or may not be compounded, they simply serve as examples of the importance of determining potency and stability. In these situations, a small error can cause significant harm to the patient. It is imperative to understand the difference between potency and stability testing so that a concerted effort can be made to meet quality standards.
Potency
Potency is defined as the concentration of the drug in a compounded preparation.5 Potency tests are known as quantitative tests and are designed to determine how much of the drug is in the sample.3 High-performance liquid chromatography (HPLC) is the method typically employed in determining potency.5 HPLC is preferred because it is very specific and efficient. Although HPLC can be used in stability-indicating methods, not all HPLC procedures are stability indicating, and they must not be assumed to be so.6 Other methods used to test potency include titration, which uses the principles of chemistry, and microbial assays, which are sometimes used to test antibiotics.5 Titration is based upon a known chemical reaction with the tested drug.5 A microbial assay uses bacteria to examine what is known as “zones of inhibition” by the antibiotic in question.5 When used alone (without chromatography), ultraviolet (UV)/visible spectrophotometry can be employed to determine potency of a single analyte in solution. In this test, multiple compounds could interfere with absorption, yielding erroneous results. When performing a potency test, the method used determines whether stability also can be determined. As already mentioned, only a stability-indicating method can be used to determine stability.
This is where some compounding pharmacists run into problems. For example, let’s say your pharmacy contracts an analytical laboratory to run a potency test on your compound and you want results at day 0, 30 days, and 60 days. The target concentration of your compound is 10 mg/mL. The test performed indicates potency only, not stability. In other words, at the predefined time points of day 0, 30 days, and 60 days, the lab analyzes only how much of the compound is present. The method used could not differentiate the compound of interest from degradants or excipients in the preparation. The results reported indicate that the concentration of the compounded preparation was 10 mg/mL at each time point. This result cannot be extrapolated as representing stability at 60 days because the preparation may have contained degradants or excipients that were present but not detected by this analysis. To put it into numbers, for example, the reported concentration of 10 mg/mL may have comprised only 6 mg/mL of the active ingredient, while 3 mg/mL was degradants and 1 mg/mL was excipients. The most important thing to recognize is that some tests determine potency but not stability. Had stability-indicating methods been used to determine potency in this case, then the results could have been used to determine a beyond-use date (i.e., stability). If a stability-indicating method showed the concentration at time 60 days to be 10 mg/mL, you could be sure that the entire 10 mg/mL was active ingredient.
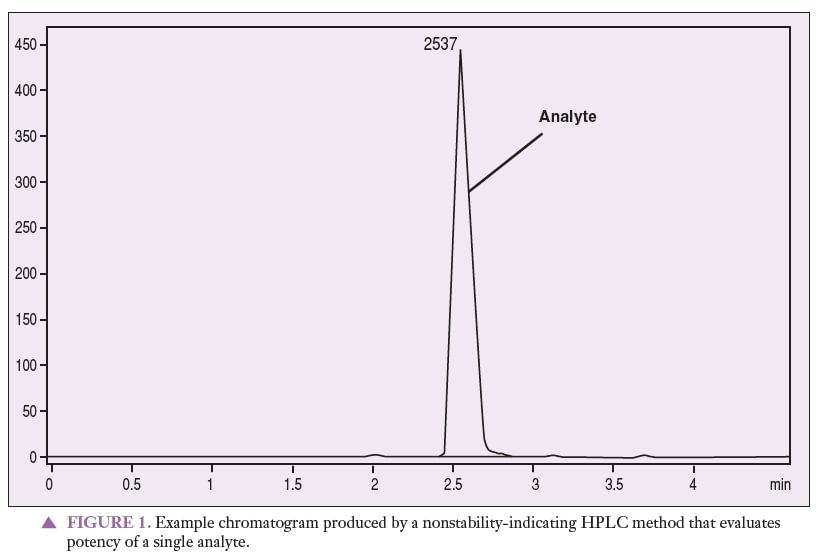
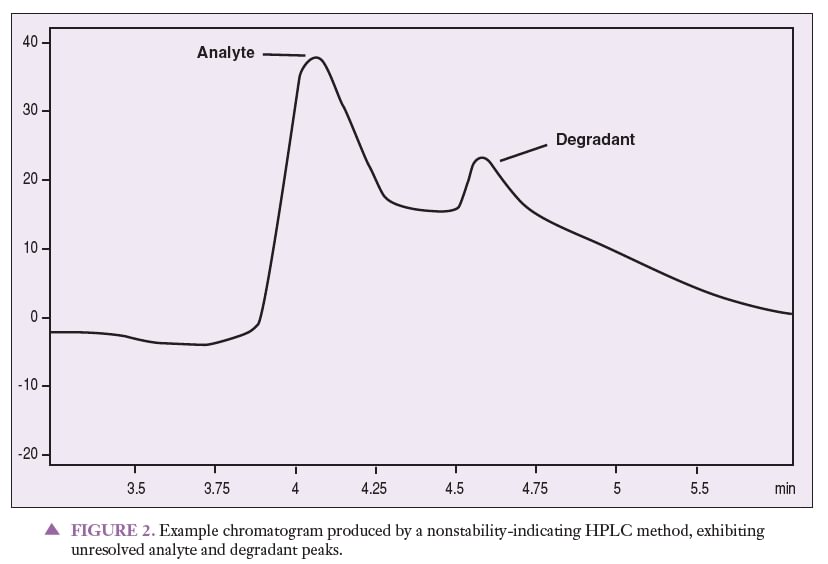
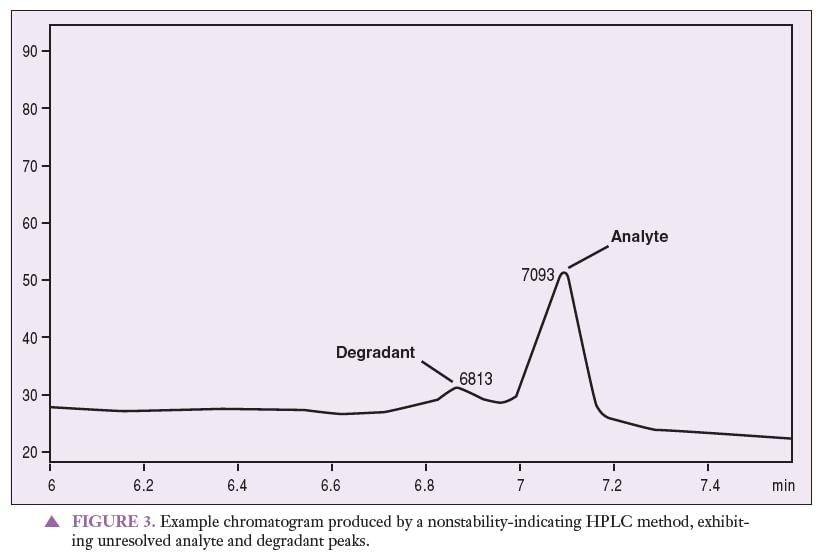
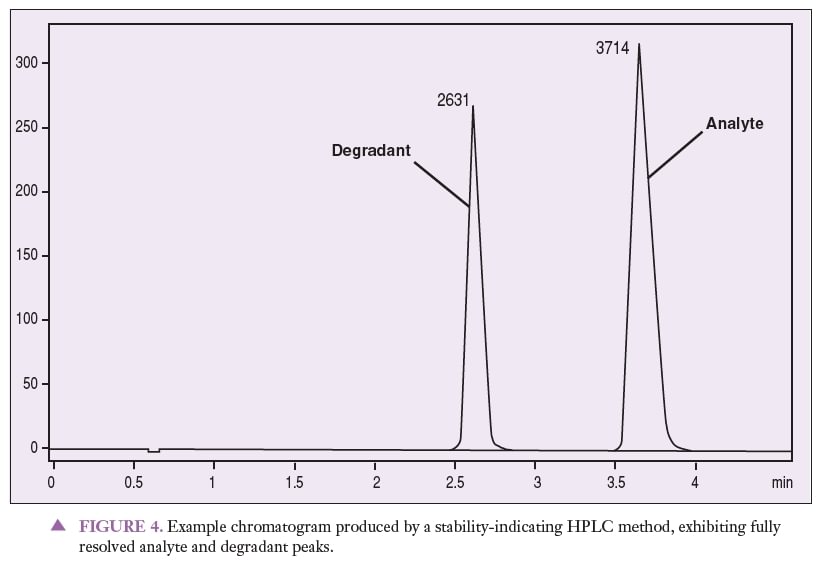
Figure 1 represents a chromatogram produced by a nonstability-indicating HPLC method that can be used to quantitate an analyte of interest. Figures 2 and 3 represent chromatograms produced by a nonstability-indicating HPLC method, exhibiting analyte and degradant sample peaks that are not resolved. All that can be concluded is that degradants were present in the sample at the time of the analysis. In Figures 2 and 3, no conclusions can be made about potency or stability. The peaks are not resolved, and thus it is impossible to properly quantitate the analyte (i.e., determine potency). Stability cannot be determined simply because stability-indicating methods were not used.
Stability
In USP Chapter <795>, stability is defined as “the extent to which a preparation retains, within specified limits, and throughout its period of storage and use, the same properties and characteristics that it possessed at the time of compounding.”2 This chapter defines beyond-use date as “the date after which a compounded preparation is not to be used and is determined from the date the preparation is compounded.”2 Stability testing is used to determine a beyond-use date, which is required by USP guidelines to be on the label or package of a compounded preparation.2 The terms stability, shelf life, and beyond-use date can be used interchangeably when referring to compounded preparations. The term expiration date is used when referring to manufactured products.
Chapter <797> Pharmaceutical Com- pounding—Sterile Preparations states, “It should be recognized that the truly valid evidence of stability for predicting beyond- use dating can be obtained only through product-specific experimental studies.”7 It is important to remember that the analytical method employed is key to determining stability versus potency. Once again, a stability-indicating method must be used to establish stability. Furthermore, a potency test that used stability-indicating methods can be used to determine stability as well as potency.
Stability testing usually includes method development, method validation, and a stability study. The method must separate the active ingredient from its degradants and impurities, as well as any other excipients in the preparation. This is done by force degrading the active ingredient and inactive ingredients to ensure that no degradants interfere with the analysis. In the process of force degradation, the compound is exposed to high heat and humidity, UV radiation, an acid, a base, and peroxide.8 It is this step that differentiates a stability-indicating test from a simple potency test. Figure 4 is an example chromatogram produced by a stability-indicating HPLC method, showing analyte and degradant peaks that are fully resolved from one another. When looking at this chromatogram, it is important to notice that the active ingredient is completely separated from its degradants. Stability can be determined from this type of study, simply because stability-indicating methods were used in the analysis.
Method validation ensures that the method meets certain criteria. The typical analytical characteristics used in method validation include accuracy, precision, specificity, detection limit, quantitation limit, linearity, range, and ruggedness, as outlined in USP Chapter <1163>. The stability study includes storing the preparation in stability chambers, testing it at predetermined time points, and then determining stability. These time points may be specified by the compounder or dictated by the particular compound. Once again, it is crucial to understand that the methods used to deter- mine stability must be stability indicating.
Quality assurance programs are essential to establishing appropriate standards for compounded preparations. The specific program implemented is up to the com- pounding pharmacy but should include a standard operating procedure, documentation, verification, and testing as outlined in USP Chapter <1163>.4 The standards of the Pharmacy Compounding Accreditation Board state that “a pharmacy must provide documentation of the basis for its determination of the beyond-use date assigned to its compounded preparation.”9
Conclusion
Employing the proper method to determine potency or stability is the key to understanding the difference between potency testing and stability testing. Methods of determining potency may or may not be stability indicating, whereas methods used to determine stability must be stability indicating. Stability-indicating methods can determine both potency and stability. It is easy to see that the madness truly lies in the methods.
References
- Trissel LA. Avoiding common flaws in stability and compatibility studies of injectable drugs.
Am J Hosp Pharm 1983; 40(7): 1159–1160.
- United States Pharmacopeial Convention, Inc. United States Pharmacopeia 27–National Formulary 25. Rockville, MD: US Pharmacopeial Convention, Inc.; 2007; 2 Supplement: 334, 511.
- Kupiec T, Huerta PL Jr. Analytical testing of extemporaneously compounded preparations. IJPC
2000; 4(2): 105–107.
- Kupiec TC. Quality-control analytical methods: Chemical testing aspects of USP Chapter <797> for compounded sterile preparations. IJPC 2005; 9(2): 136–138.
- United States Pharmacopeial Convention, Inc. United States Pharmacopeia 31–National Formulary 26. Rockville, MD: US Pharmacopeial Convention, Inc.; 2007. [In press.]
- Trissel LA. Stability studies: Five years later. Am J Hosp Pharm 1988; 45(7): 1570.
- [No author listed.] Chapter <797> Pharmaceutical Compounding—Sterile Preparations. PF 2007; 32(3): 952.
- Kupiec TC. Dr. Kupiec’s corner: What’s in a name? Beyond use dating–101, 201,301. The Pharmacists Link. Oklahoma City, OK: Analytical Research Laboratories, Inc.; Spring, Summer, Fall 2003.
- [No author listed.] Pharmacy Compounding Accreditation Board. PCAB standards with compliance indicators: Standard 6.10 beyond-use date. 2006; 7.2: 20.
Kupiec T, Skinner, R, Lanier, L. “Quality Control Analytical Methods – Stability Versus Potency Testing – The Madness is in the Method”. IJPC 2008; 12(1): 50–53
Aaron K. Thompson, Forensic & Technology Leader
Drug diversion is the illegal practice of transferring regulated pharmaceutical agents from legal sources for illicit use. Instances of drug diversion within the workplace pose a significant risk of civil liability to the employer. Not only can harm befall the drug-diverter, but contaminated drug products also pose a significant threat to patients including risk of infection, allergic response to substituted components within the drug product, and death. It is of the utmost importance for organizations to have operating procedures and surveillance programs in place which minimize the risk of drug diversion.
ARL Bio Pharma has extensive experience working with clients who face potential litigation from acts of drug diversion. In one instance, we received ten Hydromorphone syringes from a hospital. The organization wanted to know whether the syringes had been adulterated with another drug-containing product. Our analytical approach was multi-faceted. We tested the Hydromorphone potency within the syringes using traditional High Performance Liquid Chromatography (HPLC) and determined that most of the syringes contained < 5% of the expected label concentration. We further tested the samples using mass spectrometry and detected no additional compounds beyond Hydromorphone. Finally, we performed Ion Chromatography (IC) and determined that the sample concentrations were in align with that of 0.9% sodium chloride, suggesting the samples had been adulterated in normal saline.
In another high-profile case, we tested samples of Tobramycin Sulfate and Invanz (Ertapenem) that were purposefully compounded sub-potent by a pharmacist. The pharmacist had already confessed to the crime. Due to the lack of chromophore on Tobramycin, conventional HPLC-UV was not an option, Evaporative Light Scattering Detection (ELSD) was utilized as a detector. Tobramycin was only detected in one of the six submitted samples. No Ertapenem was detected in the Invanz sample.
ARL Bio Pharma is well equipped with a host of analytical instrumentation designed to test even the most complex of samples. ARL can test most commercial drug products for potency, microbiological activity, and determine the presence of “unexpected” components within samples using mass spectrometry. We issue Chain of Custody documentation to ensure the sample location and history are documented while in our presence. This is important for subsequent legal proceedings. Photographs of the sample(s) are taken, and a report generated. Our company has years of expert witness testimony in our portfolio and we are here to reveal forensic truth through sound, scientific practices. If you are in the midst of a drug diversion situation, ARL is available to talk to you about your specific situation and test your samples accordingly.
Berenice Dethier, Research Chemist II
A potency test measures the amount of API in a formulation to ensure the product is within its specifications. Criteria for acceptance are set by the client and/or follow the industry benchmarks (generally set by USP).
Several factors can contribute to a failing potency result. Investigating the root cause of the failing potency test is an important practice to characterize the true cause(s) of the failure. The goal of the out of specification (OOS) investigation is to identify what, how, and why the failure occurred. To answer all three questions, OOS investigations should include a comprehensive review of all aspects of the formulation procedure. The review typically includes everything between the acceptability of the raw materials to the validity of the potency test result. Understanding the factors that contributed to the potency failure allows the pharmacist a more strategic approach in eliminating future failures. The more specific the findings into the failure, the easier it is to properly address.
The OOS investigation first seeks to confirm the validity of the failing test result. This portion of the investigation includes a thorough review of the reference standard and sample preparations, the raw test data, the laboratory notebook write-up, test method trending, the analysts who performed the testing, and in some cases retesting of the sample to confirm the initial failing results. Once a failing potency result is confirmed, the investigation can then focus on the actual formulation process.
1. Miscalculation in the formulation
Failure to consider the molecular weight conversion or calculation mistakes are typical causes for differences between the label concentration and the measured concentration. These causes for OOS results are identified by reviewing the formulation sheets with the client. Examples of situations prone to mistakes are:
- Drugs compounded as free base vs. salt form
- Drugs containing waters of hydration
- Drugs absorbing water
For more information on how to perform calculations under these conditions, see the article about Drug Formulation.
Calculation issues can also arise when a formulation is scaled up or down. Proper training and careful review of the pharmacist’s calculations are keys to eliminate these errors.
2. Improper or incomplete labeling
OOS results have been encountered when samples were labelled incompletely. Cases we have come across at ARL Bio Pharma include Amino Acids: the label indicated Lysine, the samples were out of specifications, but after looking at the formulation sheet we discovered that the concentration was calculated for Lysine HCl, and the samples were passing with the appropriate calculations. Other common situations are Fentanyl compounded from Fentanyl Citrate and Morphine Sulfate from Morphine Sulfate Pentahydrate. In these cases, we follow USP guidelines for reporting and OOS samples are rare.
Other problematic situations include labels stating a percentage with no other qualifiers, which is ambiguous: is it volume per volume, weight per volume or weight per weight concentration? This is typical for creams/ointments and for liquid active ingredients such as antimicrobial agents. Ointments are sampled by weight and solutions by volume. Consistency between how the sample amount is measured and how the concentration is expressed greatly decreases approximations on density and calculation mistakes, and avoids OOS results.
When the active ingredient is added to IV bags, the concentration should be expressed per bag rather than per volume to avoid OOS results, since adding solution increased the fill volume of the bag. For example, a sample labeled as “Vancomycin 1 mg/mL” can be prepared by adding 10 mL of 100 mg/mL Vancomycin to a 1000 mL bag, which may have a 10% overfill. This results in an actual concentration of 0.9009 mg/mL. In such a case the potency is more appropriately expressed as 1 g of Vancomycin per bag where the fill volume of the bag has been determined.
The labels should be self-explanatory and analysts should not need the formulation records to understand what they are working with.
3. Improper compounding procedure
Oversights in compounding can affect potency at multiple levels. A trained pharmacist and a thorough Quality System should eliminate most sources of error.
A regular cause for potency failures stems from the mixing of triturated powder blends. ARL Bio Pharma often performs potency testing on triturated powder blends at 0.1% w/w or lower. At these concentrations, achieving a homogenous blending can be challenging. Out of Specification results are usually investigated hand in hand with the client (after intra-laboratory investigation has ruled out sample preparation or method problems). Often additional samples or aliquots are tested. If the sample results are sometimes high and sometimes low, we can conclude there is a lack of uniformity in the blend. Content uniformity also affects suspension formulations and creams/ointments should phase separation occur over time. In other instances, the results are consistent but sub-potent indicating loss of API during the compounding process (for example issues with static electricity causing binding of the API to equipment).
More cases where compounding was the culprit in the OOS investigation include:
- Container closure issues (example: an Alprostadil solution in Alcohol improperly closed, causing the samples to be super potent when the concentration increased upon solvent evaporation)
- Improper handling or storage of drug causing the API concentration to be low (exposure to moisture, light, etc.)
- Improper choice of filters: non-specific binding can occur, we recommend to test potency before and after filtration during formulation development
Successful potency out of specification (OOS) investigations identify why the sample did not pass. These investigations often require collaboration between the testing laboratory and the compounding pharmacy. If the two groups do not work together, assumptions are made and a root cause is typically not identified, resulting in repeated OOS test results. ARL Bio Pharma has years of experience in this collaborative approach and is happy to assist in troubleshooting complicated compounding procedures, upfront or following potency failures.
ARL now offers USP <1207> container closure integrity testing (CCIT) on IV bags, syringes, cassettes, and vials.
Three types of leaks that can be detected during CCIT include:
- entry of microorganisms
- escape of the product dosage form or entry of liquids or solids
- escape of nitrogen gas or entry of oxygen, water vapor, or air gases
Historically, CCIT has been performed using probabilistic tests such as dye ingress or microbial immersion. Recently, USP issued new guidance requiring deterministic tests to achieve more reproducible and predictive results. While ARL will continue to offer probabilistic dye ingress testing, our new vacuum decay test method meets USP’s requirements for a deterministic test.
The vacuum decay method:
- can be used to test all types of container closure systems
- tests for gas leaks
- tests for liquid leaks
- can provide a significant savings to our clients because the test articles can be used for other tests during a stability study
- provides a high level of sensitivity (down to 5 micron), repeatability, and accuracy with a PASS / FAIL result
The method is performed by placing a package into a test chamber, drawing a vacuum on the test chamber, and monitoring it for any changes in the vacuum level. If the package is defective, air, drug product, or vapor will leak from the package into the test chamber, causing a change in pressure.
Properly sealed packages do not leak anything into the chamber, holding the vacuum level constant.
For more information on CCIT, contact ARL at 800-393-1595 or info@arlok.com.
Vince Bobin, Laboratory Director
Some of the same challenges in creating a formulation are also related to getting accurate and representative results from your Analytical Testing. This article outlines how to avoid some common issues to help ensure the quality of compounded products.
Topics include:
- Drugs as free base versus salt forms
- Drugs containing waters of hydration
- Drugs absorbing water
- Drug content uniformity
Salt vs. Free Base
Many pharmaceutical drugs are manufactured as salts such as citrate or hydrochloride. A common example is Fentanyl Citrate where Fentanyl is the free base and Citrate is the salt. When formulating or testing a compounded preparation it is critical to know if the labeled concentration is referring to the free base or the salt form.
In USP the Fentanyl concentration is shown as the free base. The weight of the salt has to be factored into the formulation to get the correct concentration of Fentanyl only. To make 1 mL of 50 mcg/mL Fentanyl you would need to weigh out 78.5 mcg of Fentanyl Citrate.
Molecular weight of Fentanyl Citrate = 528.59
Molecular weight of Fentanyl = 336.48
50 mcg (as Fentanyl) x 528.59/336.48 = 78.5 mcg (as Fentanyl Citrate)
In this example if the formulation had not corrected for the salt and had only contained 50 mcg of Fentanyl Citrate the potency would have only been 64% of the intended concentration. It is extremely important to be clear and specific about what the labeled concentration refers to.
For other APIs like peptides such as GHK-Cu Acetate, there are multiple forms that can be reported as: GHK, GHK-Cu, GHK Acetate or GHK-Cu Acetate. It is imperative that during the compounding process, the calculations performed for material needed, the right values are being accounted for. These values are provided in the CofA from the manufacturer.
Waters of Hydration
Similar to salt forms, some drugs contain waters of hydration or also known as ‘hydrates’. They contain water as part of their chemical structure. An example is Morphine Sulfate Pentahydrate which is both a salt and hydrate. Water is often ‘corrected for’, like the salt form example above, and additional drug must be weighed out to account for the weight of the water. For some hydrates this may not be correct as USP defines Morphine Sulfate Compounded Injection as the concentration of morphine sulfate pentahydrate which includes the water. If the waters of hydration had been ‘corrected for’ the preparation would have been super potent.
Water Absorption
Many drugs contain water that is not part of its chemical structure. That water is often referred to as free water or absorbed water. Some drugs, such as Betamethasone Sodium Phosphate, readily attract and absorb water and are referred to as hygroscopic. Betamethasone Sodium Phosphate in USP has a limit for water at 10.0%. If the vendor certificate of analysis listed the water content as 8.5% this water can be corrected for in the formulation by weighing out additional drug.
However, if a single container was opened and closed to make several batches the drug is likely to absorb additional water. If enough water is absorbed it can cause potency failures since the amount of water being corrected for is now much less than the actual amount of water the Betamethasone Sodium Phosphate contains.
If water absorption is suspected to be the cause of a low potency result the drug can be sent in for water testing. Testing is performed either by Karl Fischer (specific for water) or Loss on Drying (general) depending on the drugs specific USP monograph.
Content Uniformity
Generally applied to individual dose forms such as tablets and capsules. Content uniformity refers to how even the amount of drug contained in each individual unit is. Variation in the amount of drug in a capsule, for example, can come from many different steps during the process
- Blending of the drug with excipients
- Segregation of the drug during capsule fill
- Amount of blend added to the capsule
If potency results on a batch are coming back high or low testing individual dosages from the beginning, middle and end of the process can help to determine if uniformity is the cause. If variability is observed additional testing can be performed at each step of the compounding process, e.g., top, middle, bottom of the blend, to identify where the variability is coming from and fix it.
While often considered for tablets and capsules the same principles can be applied to other dose forms as well. Proper mixing is critical for creams and ointments as well as good formulation development to prevent phase separation during shipping and over time.
Individual vials of suspensions may contain a consistent total volume but can vary in the amount of suspended drug if proper mixing/filling does not occur.
Content Uniformity is important to remember when assessing low or high potency results. It should not be assumed that an incorrect amount of drug was added to the batch. It may be present just not uniformly distributed.
Conclusion
During formulation and analytical testing be specific when referring to the concentration of the drug. Does it refer to the salt form or the free base? Should the amount of drug be corrected for water or not? If a potency failure is obtained during testing, first ensure you are comparing ‘apples to apples’ and the drug/amount are correct for your specific formulation. Also consider if the drug may have absorbed water (low potency) or if there may be content uniformity issues (low or high potency).
Analytical testing can help troubleshoot these common formulation problems and establish the quality of the compounded preparation.
Please contact ARL for more information 800-393-1595 or info@arlok.com.