
Nicholas Pryor, ARL Bio Pharma Associate Lab Supervisor, Microbiology
Understanding USP 71 Method Suitability
Sterility testing by USP 71 is performed in conjunction with other sterility assurance procedures to help ensure that sterile preparations are free of microbial contamination and safe for patient use. Before sterility testing can be reliably performed on a product, a method suitability test must first be completed for the specific formulation.
What is USP 71 Method Suitability?
USP 71 focuses on sterility testing, and method suitability is a critical requirement within this framework. Method suitability demonstrates that the sterility test method used is capable of reliably detecting viable microorganisms in a specific pharmaceutical formulation.
In simple terms, method suitability answers the question:
“Can this sterility test reliably detect microorganisms in this specific product formulation?”
To verify this, known microorganisms are intentionally introduced into the product under controlled conditions. The test method must then successfully recover and support the growth of these microorganisms. If recovery cannot be demonstrated, the method is not considered suitable for that formulation.
Method suitability is formulation-specific because inhibitory properties can vary significantly between products. Active ingredients, inactive ingredients, preservatives, and vehicles may all interfere with microbial recovery.
Why Does Method Suitability Matter?
Pharmaceutical formulations can interfere with sterility testing in multiple ways. Without proper method suitability validation, there is a risk of false-negative sterility results, where a contaminated product may incorrectly appear sterile.
Common formulation challenges include:
- Antimicrobial preservatives that inhibit microbial growth
- Antibiotics or active ingredients with antimicrobial activity
- Viscous or oily formulations that trap microorganisms
- Suspensions or poorly soluble products that are difficult to filter
- Extreme pH or excipient interactions that suppress microbial recovery
Method suitability ensures that these formulation-specific challenges are identified and addressed before routine sterility testing is performed.
How is Method Suitability Demonstrated?
A sterility test method is selected based on a detailed evaluation of the product formulation and any applicable sub-formulations. The two primary sterility testing approaches described in USP 71 are closed-membrane filtration and direct inoculation.
Closed-Membrane Filtration
Closed-membrane filtration is the preferred sterility testing method because it separates the product from potential microbial contaminants, helping reduce or eliminate inhibitory effects from the formulation.
During the test:
- Product is passed through sterile closed-membrane filtration systems.
- Microorganisms are retained on the membrane filters while the product is directed to waste.
- The membranes are rinsed using validated rinse fluid to remove residual product inhibition.
- Following rinsing, growth media is aseptically added directly into the closed-membrane filtration systems, including:
- Fluid Thioglycollate Medium (FTM/FTG)
- Tryptic Soy Broth (TSB)
The closed systems containing the membrane filters and growth media are then incubated and monitored for evidence of microbial growth.
Direct Inoculation
Direct inoculation is typically used for products that cannot be filtered, such as certain oils, suspensions, or highly viscous formulations. In this method, the product is directly diluted into the growth media to reduce inhibitory effects. Additional neutralization strategies may also be required.
Compendial Test Microorganisms
USP 71 method suitability studies utilize six compendial challenge microorganisms that represent a range of bacterial and fungal microorganisms commonly associated with manufacturing environments and contamination risks.
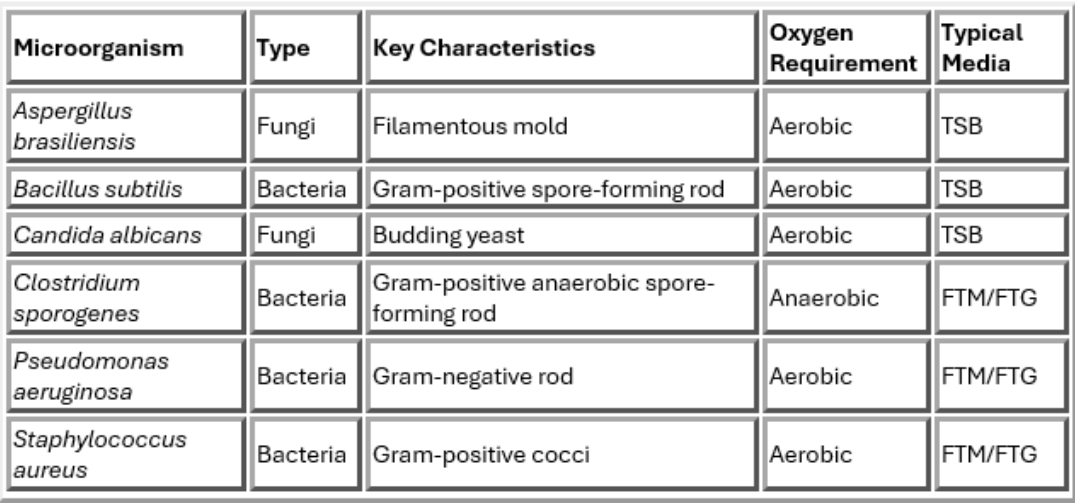
The method suitability test is performed by inoculating the selected media with less than 100 colony forming units (CFU), and confirming successful recovery and growth.
What Happens if Recovery Fails?
If all challenge microorganisms are not successfully recovered, the method must be modified and re-evaluated. Additional neutralization strategies may include:
- Increasing product dilution
- Adding validated neutralizers
- Incorporating pre-filtration dilution steps
- Performing additional membrane rinses
- Selecting alternative test methodologies
Increasingly conservative approaches may be required until all six challenge microorganisms can be adequately recovered.
Regulatory Importance
Demonstrating method suitability is a regulatory expectation and a critical component of sterility assurance programs. Regulatory agencies, including the FDA and international guidance documents such as ICH, expect manufacturers and testing laboratories to demonstrate that sterility testing methods are effective for each specific formulation.
Failure to establish method suitability may:
- Invalidate sterility test results
- Delay product release or approval
- Trigger regulatory observations or compliance concerns
- Increase patient safety risk
Importantly, the validated method remains applicable only as long as the formulation remains unchanged. Changes to active ingredients, preservative systems, vehicles, suppliers, concentrations, or sub-formulations may require method suitability to be repeated.
Key Takeaway
USP 71 method suitability is essential for:
- Ensuring sterility test accuracy
- Detecting formulation-specific inhibition
- Adapting sterility methods to challenging products
- Supporting regulatory compliance
- Protecting patient safety
By confirming that sterility testing methods can reliably recover viable microorganisms in a specific formulation, pharmaceutical manufacturers and testing laboratories can maintain confidence that sterility results are meaningful, accurate, and protective of patient health.
For more information on method suitability, contact ARL today and start the process today to obtain meaningful, accurate sterility results that prioritize patient health.

Amber Gilbert, ARL Bio Pharma Technical Sales Representative
API Vendor Qualification is a critical component of quality assurance for compounding pharmacies and outsourcing facilities. A structured, risk‑based qualification program helps ensure vendor reliability, regulatory compliance, and most importantly, patient safety across the supply chain.
Why API Vendor Qualification Matters in Pharmaceutical Compounding
In pharmaceutical compounding, product quality starts with the supply chain. The identity, purity, and consistency of active pharmaceutical ingredients (APIs), excipients, and packaging components directly affect the safety and performance of the final preparation. A strong vendor qualification program helps control supply chain risk, support regulatory compliance, and protect patient safety.
Vendor qualification is a formal, documented process used to evaluate, approve, and monitor suppliers and manufacturers of critical materials against defined quality and regulatory expectations. Not all materials and not all vendors carry the same level of risk. As a result, vendors should be selected and managed through established quality system controls using a risk-based approach.
Key considerations in API vendor qualification often include:
- Material Quality and Conformance: Identity, purity, and lot-to-lot consistency
- Patient Safety Risk: Control of impurities, contaminants, and suitability for intended use
- Supply Reliability: Production/Distribution capacity, lead times, and historical quality performance
Vendors that cannot consistently meet these expectations may increase the likelihood of deviations, impact formulation stability, or supply chain disruptions.
Meeting Regulatory Expectations
A robust vendor qualification program also supports compliance with expectations of regulatory authorities and quality standards organizations, including the U.S. Food and Drug Administration (FDA), the United States Pharmacopeia (USP), the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), and the International Organization for Standardization (ISO).
Under 21 CFR 211, supplier qualification is addressed through validation and quality system activities and supported by documented criteria such as supplier assessments or checklists. USP 1083 outlines core principles for identifying, approving, and monitoring suppliers of materials, packaging components, and services. ICH Q10 further integrates supplier management into the pharmaceutical Quality Management System (QMS) framework based on ISO 9001 concepts.
For API suppliers, it’s important to distinguish between the manufacturer and the distributor and understand expectations for each. Under ICH Q7 and FDA Q7, both must be qualified. Regulators also expect any entity performing GMP-critical activities in the API supply chain to be qualified and audited, including distributors that act as contract manufacturers/packagers or that store, handle, or transport API.
While regulations do not prescribe a single method for qualifying vendors, regulatory authorities expect pharmaceutical manufacturers, distributors, and compounding pharmacies to demonstrate due diligence in evaluating and maintaining their supply chains. Inadequate oversight can lead to FDA Form 483 observations, warning letters, product recalls, regulatory delays, and loss of consumer trust.
Essential Qualification Activities
An effective vendor qualification program typically includes several key activities:
- Vendor Selection: The process begins with identifying potential suppliers and assessing their capabilities, certifications, and regulatory compliance history.
- Vendor Audit & Risk Assessment: Each vendor should be evaluated based on material criticality and supplier risk to determine the appropriate level of qualification and oversight. Evaluation may include quality system assessments, documentation review, regulatory history, on‑site or remote audits, and laboratory testing of representative samples.
- Documentation & Compliance: Qualified vendors should provide key documents such as quality agreements, Certificates of Analysis (CoAs), stability data, and safety data sheets (SDS). When a CoA is issued by or for a repacker/reprocessor, agent, or broker, it should identify the testing laboratory (name, address, and phone number) and reference the original manufacturer and original batch CoA; a copy of the original batch CoA should be attached. Approval or disqualification decisions must be documented, and records retained per quality system requirements.
- Ongoing Monitoring and Requalification: For qualified vendors, written quality agreements establish expectations for documentation review, change control, communication, and performance. Ongoing monitoring may include periodic audits, questionnaires, interviews, and routine analytical testing of incoming materials against predefined acceptance criteria.
Vendor qualification is not a one‑time event, but a continuous quality and compliance control designed to ensure upstream suppliers consistently meet established requirements. By maintaining effective supplier oversight, compounding pharmacies can better manage risk while meeting regulatory expectations and continue to deliver safe, high-quality preparations to patients.
How ARL Can Help
ARL Bio Pharma supports vendor qualification programs through comprehensive testing, documentation review, and quality‑focused expertise. To learn more about how ARL can support your supplier qualification and material testing needs, contact info@arlok.com or call (800) 393‑1595.
Resources
- 21 CFR 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals
- United States Pharmacopeia 1083 Supplier Qualification
- FDA Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients, Guidance for Industry
- ICH Harmonised Tripartite Guideline Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients Q7
- ICH Harmonised Tripartite Guideline Pharmaceutical Quality System Q10
- ISO 9001:2015 Quality Management Systems – Requirements

Tiffany Corbin, ARL Bio Pharma Associate Lab Supervisor, Microbiology
Compounding pharmacies and outsourcing facilities must ensure that sterile preparations are compounded in controlled environments to minimize the risk of microbial contamination. Environmental monitoring (EM) is a process required by the United States Pharmacopeia (USP) and the Food and Drug Administration (FDA) to verify that aseptic processing areas consistently maintain a state of control during compounding. Pharmacies and facilities are required to document their EM procedures, including the selection of sampling sites, the frequency of sampling, acceptance criteria for monitoring particulates and microorganisms, and the investigative and corrective actions taken when necessary.
Sampling Sites
When selecting sampling sites, it is important to consider both routine operations and worst-case scenarios. The selection of these sites should be based on a documented risk assessment that accounts for airflow patterns, room classifications, personnel activities, equipment placement, and proximity to sterile preparation areas.
Common sampling locations typically include:
- Critical sites within ISO environments
- Equipment surfaces and high-touch areas
- Cleanroom floors, walls, and pass-throughs
- Personnel gloves and gowns following aseptic manipulations
Sampling should occur under dynamic conditions, with operations actively ongoing, to yield relevant data regarding routine processing conditions. Relying solely on static conditions for sampling does not adequately demonstrate environmental control.
Worst-case scenario conditions may include periods of maximum personnel occupancy, high batch throughput, upper/lower limits of temperature or humidity, and differential pressure excursions (within acceptable action limits).
Sampling Frequency
The FDA and USP specify sampling intervals based on the ISO classification of the area and the category of compounded sterile preparations. More frequent monitoring is required during initial qualifications, after any significant changes, or when adverse trends or excursions are detected. Additionally, it is important to consider when samples are collected, particularly during peak activity periods and at the conclusion of operations.
Sample Collection
Various methods are used to collect EM samples, each serving a specific purpose in assessing environmental conditions. Using a combination of these sampling methods allows for a comprehensive evaluation of the environment:
- Active air sampling draws a measured volume of air over a growth medium to quantify viable airborne microorganisms and is a requirement in classified areas.
- Passive air sampling (using settle plates) serves as a supplementary method in ISO environments.
- Surface sampling involves contact plates (e.g., RODAC plates) or sterile swabs to evaluate microbial contamination on surfaces and is essential for routine monitoring.
All sampling media must promote microbial growth and have successfully passed growth-promotion testing prior to use, in accordance with USP requirements.
Incubation and Observation
After collecting EM samples, they must be incubated under controlled conditions to allow for microbial growth. USP and FDA recommend a two-stage incubation approach using temperature ranges of 20-25°C and 30-35°C to ensure recovery of both environmental and human-associated microorganisms, including slow-growing microorganisms.
Once the incubation is complete, the samples are examined for microbial growth. Pharmacies and facilities should compare the results against established acceptance criteria, including alert and action levels specific to the ISO environment. It is essential to establish alert and action levels based on regulatory guidance, historical data, and risk assessment. These alert and action levels serve as indicators to identify potential issues or prompt further investigations, rather than simply categorizing results as pass or fail.
Interpreting Results and Investigations
It is important to trend and review EM results. USP emphasizes that trends observed over time are often more meaningful than individual data points. Gradual increases in microbial recovery or changes in microorganism types can signal a potential loss of control.
When results exceed established limits or show adverse trends, pharmacies and facilities must initiate an investigation that includes:
- Identifying the recovered microorganism down to the genus and, when applicable, species level.
- Assessing potential sources of contamination, including personnel practices, cleaning procedures, or equipment issues.
- Reviewing recent actions or interventions that may have contributed to the contamination.
- Evaluating environmental monitoring trends to determine whether the issue is an isolated incident or part of a broader pattern.
The goal of EM goes beyond simple detection; it aims to promote continuous improvement and maintain control in aseptic processing environments. Ongoing evaluation, trending, and refinement of the EM program are vital for safeguarding the safety, quality, and sterility of compounded preparations.
ARL Bio Pharma Testing Services
ARL Bio Pharma provides incubation and enumeration testing for EM samples. Pharmacies and facilities can send plates to ARL for incubation, colony-forming unit (CFU) counting, and a Certificate of Analysis to document the results for EM programs. To demonstrate microorganism growth capabilities, ARL offers growth promotion testing of media used in EM programs, media fills, and personnel qualifications. Additionally, microbial identification testing is available to identify microbial contaminants and support investigative and corrective actions to improve compounding processes.
For more information on ARL testing services, contact info@arlok.com or 800-393-1595.
Resources:
- USP 797 Pharmaceutical Compounding—Sterile Preparations
- USP 1116 Microbiological Control and Monitoring of Aseptic Processing Environments
- FDA Guidance for Industry – Current Good Manufacturing Practice – Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act
- FDA Guidance for Industry – Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice