Bailey Rubin, ARL Bio Pharma Technical Sales Representative
Analytical testing plays a critical role in ensuring that compounded preparations meet established quality, safety, and regulatory standards throughout the preparation’s lifecycle, including development, stability testing, and final release. One primary way this is achieved is by using validated analytical methods, which are formulation-specific and demonstrate that the test procedure produces reliable, accurate, and reproducible results for its intended purpose.
Organizations such as the U.S. Food and Drug Administration (FDA), the United States Pharmacopeia (USP), and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) publish guidance and standards that define expectations for pharmaceutical testing. According to ICH Q2(R2) Guideline on Validated Analytical Procedures, the analytical performance characteristics to be evaluated depend on the type of test procedure and its intended purpose. The typical validation parameters for analytical methods used to measure specific attributes in release, stability or impurity testing include:
- Accuracy
- Precision
- Linearity and Range
- Specificity
- System Suitability, if measurements are susceptible to various analytical conditions
- Detection Limit
- Quantitation Limit
- Robustness
Together, these characteristics confirm that the method can accurately measure the active ingredient in a formulation while accounting for other components such as excipients, impurities, or potential degradation products.
Validated analytical methods are critical for supporting release, stability, and impurity testing of compounded preparations. For example, potency assays used for quality control release testing must accurately measure the active pharmaceutical ingredient (API) in the finished dosage form, while stability studies require stability-indicating methods capable of separating the API from excipients and potential degradation products to monitor changes over time.
Regulatory expectations for validated methods vary depending on the type of compounding facility. 503B outsourcing facilities, which operate under current good manufacturing practice (cGMP) regulations (21 CFR Parts 11, 210–211), are generally expected to use validated analytical methods for all routine release and stability testing. 503A compounding pharmacies, which operate under a different regulatory framework, are not strictly held to cGMP requirements; however, analytical methods used to support extended beyond-use dating (BUD) or stability claims should still be demonstrated to be suitable for their intended purpose (USP 795 Pharmaceutical Compounding—Nonsterile Preparations; USP 797 Pharmaceutical Compounding—Sterile Preparations).
At ARL, our analytical team supports both 503A compounding pharmacies and 503B outsourcing facilities by developing and applying validated, stability-indicating methods to generate reliable data for stability studies, cGMP release testing, and impurity analysis, supporting the quality and stability of compounded preparations.
For questions regarding obtaining or validating an analytical method for quality control testing, please contact ARL at info@arlok.com or call 800-393-1595.
Resources:
- FDA Guidance for Industry – Current Good Manufacturing Practice – Guidance for Human Drug Compounding Outsourcing Facilities Under Section 503B of the FD&C Act
- ICH Q2(R2) – Validation of Analytical Procedures
- USP 795 Pharmaceutical Compounding – Nonsterile Preparations
- USP 797 Pharmaceutical Compounding – Sterile Preparations
- USP Formulation and Stability Reference Document for Pharmaceutical Compounding
- USP 1225 Validation of Compendial Procedures
Michael Darnaby, ARL Bio Pharma Technical Sales Representative
Visual inspection is an essential quality-control test that helps ensure patient safety and is critical to the release of finished compounded sterile preparations (CSP). Visual inspection is expected for all sterile injectables and should include evaluation of particulates, container defects, color, and clarity. Visible particulates can originate from many sources in the compounding process, including personnel and equipment. It is imperative that visual inspection results are used not only for CSP release, but also to evaluate the drug compounding process itself.
USP 797 states that 100% of the produced batch must be visually inspected. USP 1790 states that visual inspections should be performed immediately after compounding and before labeling to easily detect defects. This inspection can be performed manually or using automation. Once the 100% inspection is complete, acceptance sampling and testing should be conducted on a statistically valid sample taken from the accepted units via manual inspection. This acceptance sampling and testing should be conducted via manual inspection means only by trained personnel and under tightly controlled conditions:
- Use of contrasting backgrounds (white/black)
- Controlled light amount (2000-3750 lux)
- No magnification
- Sufficient inspection time
- Limiting inspection sessions to reduce inspector fatigue
Acceptance limits, test flow, and sampling procedures should be risk-based.
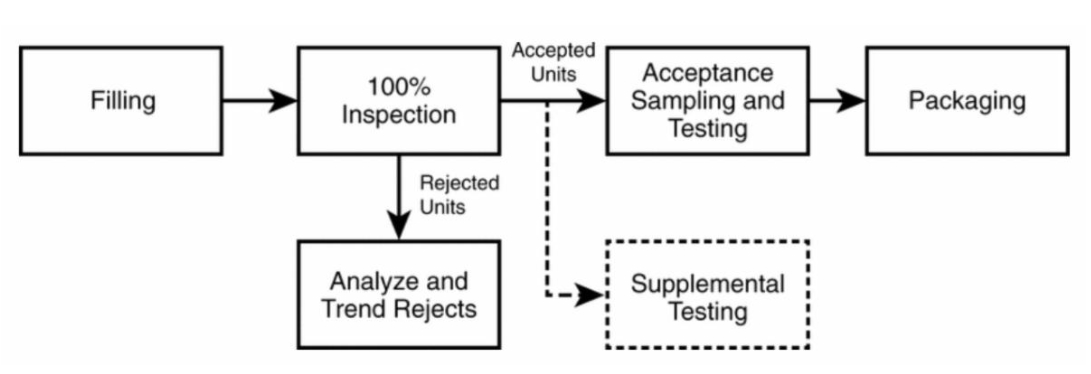
USP 1790 – Figure 1: Typical Process Flow Chart. This chart shows a simplified process flow where solid boxes indicate required process operations, and those with dotted lines (supplemental testing) may be required for difficult-to-inspect products.
For more information on establishing these parameters, please refer to USP 1790.
Visual inspection is not merely a regulatory requirement—it is critical to patient safety control. Data obtained during inspection can also help pharmacies identify process improvements, such as:
- Enhanced filtration techniques
- Improved material handling practices
- Identification of recurring contamination sources
- Refinement of aseptic technique training
Trending visual inspection failures can serve as an early warning indicator of process drift or environmental control issues.
For compounding pharmacists, visual inspection is a critical step in mitigating risk before sterile preparations reach patients. USP 790 establishes the non-negotiable expectation that injectable CSPs must be essentially free of visible particulates, while USP 1790 provides the operational framework for building reliable, consistent, and defensible inspection programs. By implementing structured inspection procedures, investing in personnel training, and incorporating ongoing performance monitoring, compounding pharmacies can significantly reduce the risk of contamination and reinforce their commitment to patient safety and CSP quality.
Resources:
- USP 1 Injections and Implanted Drug Products (Parenterals) -Product Quality Tests
- USP 790 Visible Particulates in Injections
- USP 797 Pharmaceutical Compounding – Sterile Preparations
- USP 1790 Visual Inspection of Injections
- ISO 2859-1: Sampling Procedures for Inspection
- ANSI/ASQ Z1.4: Sampling Procedures and Tables for Inspection by Attributes
- FDA Guidance for Industry – Inspections of Injectable Products for Visible Particulates